


Caso clínico
Mujer de 42 años, con antecedentes de quistes de ovario, que acude a la consulta por presentar astenia de 8 meses de evolución, lumbalgias ocasionales y un episodio aislado de disuria y hematuria con coágulos en relación con el esfuerzo.
Entre los antecedentes familiares destaca que su madre falleció por uremia a los 39 años.
En la exploración física encontramos a la palpación abdominal masas múltiples, más voluminosas en el hemiabdomen derecho, y hepatomegalia.
En la analítica destacan: hemoglobina 11,8 mg/dl, hematócrito 35%, creatinina 2,1 mg/dl, BUN 43 mg/dl, CCr 5,1 ml/min, IgA 154 mg/dl, orina elemental y urocultivo normales y proteinuria de 24 h negativa.
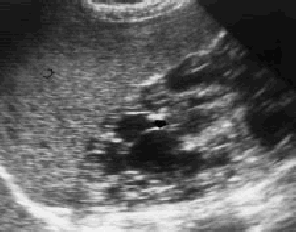
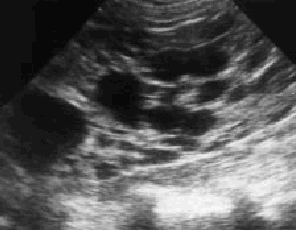
Se solicita ecografía abdominal en la que se observan múltiples imágenes quísticas renales (figs. 1 y 2) con desestructuración del parénquima y pérdida de diferenciación entre corteza y médula renal. Por todo ello se efectuó el diagnóstico de enfermedad poliquística hepatorrenal (EPHR)
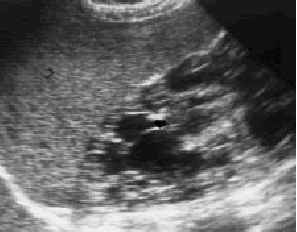
Fig. 1. Múltiples imágenes quísticas en el riñón derecho, de diferentes tamaños.
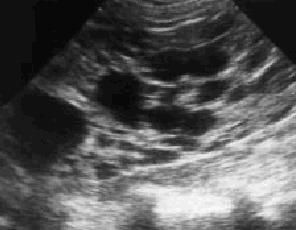
Fig. 2. Quistes renales múltiples con desestructuración del parénquima y pérdida de diferenciación entre corteza y médula renal.
Comentarios
La EPHR es una enfermedad de herencia autosómica dominante, causada por una mutación del cromosoma 16p, que cursa con riñones grandes, de superficie abollonada, y parénquima renal invadido por quistes llenos de líquido y de tamaño variable (hasta 5 cm) que se desarrollan como expansiones saculares de los túbulos renales, siendo el parénquima renal entre los quistes normal1,2.
Es una enfermedad sistémica que afecta a múltiples órganos; aparecen quistes hepáticos en el 20-25% de los pacientes y con menor frecuencia en páncreas, bazo, tiroides, ovario, endometrio, vesículas seminales y epidídimo. En un 10-20% se encuentran aneurismas en las arterias cerebrales.
Clínicamente, el inicio suele producirse entre los 30-40 años y se presenta como dolor lumbar en un 30% de los casos, hipertensión arterial (HTA) en un 21%, infección urinaria en un 19% y masas abdominales palpables en un 15%3.
En el transcurso de la enfermedad es frecuente el desarrollo de HTA (64%), infecciones urinarias de repetición (44%) y litiasis renal4.
El pronóstico empeora en los casos de desarrollo de los síntomas a edades tempranas, HTA, proteinuria y riñones palpables5.
El diagnóstico se basa en la ecografía renal y el diagnóstico de certeza se realiza cuando existe historia familiar de enfermedad y quistes renales bilaterales.
En individuos sin historia familiar se puede realizar el diagnóstico cuando se presentan dos o más de los siguientes hechos: crecimiento renal, tres o más quistes hepáticos, aneurisma de las arterias cerebrales o quistes solitarios en la glándula pineal, aracnoides, páncreas o bazo.
El diagnóstico diferencial incluye: enfermedad renal quística, neoplasias renales, pielonefritis crónica e hidronefrosis bilateral.
Es una enfermedad progresiva que evoluciona a la insuficiencia renal crónica terminal. No existe tratamiento específico, salvo el de las complicaciones.
Las dietas con restricción de proteínas son útiles para minimizar el impacto de la hipertensión intrarrenal y la glomeruloesclerosis progresiva. Cuando se alcanza el estadio de nefropatía terminal, la diálisis o el trasplante renal constituyen las únicas alternativas6-8.
La mayoría de los aneurismas cerebrales son asintomáticos; se debe considerar la intervención quirúrgica si son sintomáticos y/o mayores de 1 cm.
Los quistes hepáticos se desarrollan relativamente tarde y no contienen bilis. En las mujeres suelen ser de peor pronóstico y en ocasiones requieren aspiración, escleroterapia con alcohol o hepatectomía parcial.
Si hay dolor renal se puede realizar la cirugía reductora del quiste, que ha probado ser relativamente segura sin comprometer la función renal.
Es importante el consejo genético. Se recomienda examinar a los familiares de primer grado mayores de 20 años mediante ecografía renal y, si es negativo, valorar la realización de una tomografía axial computarizada (TAC).