






No siempre es posible establecer el diagnóstico de un recién nacido con defectos congénitos. En general esto se debe a varias causas fundamentales: a) porque el conjunto de manifestaciones clínicas que presentan son aún de causa desconocida; b) porque los síndromes que ya se conocen tienen una frecuencia tan baja que no son fáciles de reconocer cuando no se tiene experiencia previa; c) porque no existe especificidad alguna entre una causa y un defecto; d) porque algunos solo presentan rasgos dismórficos que solo con gran experiencia pueden ser evaluados, ya que la mayoría de esos rasgos se encuentran en la población general sana, y e) porque muchos de ellos son de aparición evolutiva siendo de apariencia normal al nacimiento. Sin embargo, hoy se sabe que algunos de estos síndromes son producidos por alteraciones debidas a pérdidas de porciones cromosómicas tan pequeñas que no se detectan por citogenética de alta resolución (son crípticas para estas técnicas) o a mutaciones conocidas de ciertos genes.
Como muchos de estos niños van a ir presentando sus manifestaciones durante la infancia, van a ser atendidos por los médicos de atención primaria, por lo que es esencial que dispongan de unas pequeñas guías para su reconocimiento y su manejo adecuado.
En este artículo se explican las diferentes formas de presentación, sus características, las técnicas con las que se pueden diagnosticar, así como los síntomas que pueden alertar sobre estos síndromes y forma de actuar. Porque de la identificación precoz depende mucho el pronóstico y la información a la familia.
It is not always possible to establish the diagnosis of a newborn with congenital defects. In general, this is due to several main reasons: a) because the group of clinical signs that they show are still of an unknown cause; b) because the already known syndromes are such a low frequency that they are not easy to recognise when there is no previous experience; c) because there is no specificity between a cause and a defect; d) because some only present with dysmorphic characteristics that can only be assessed by someone with wide experience since the majority of these characteristics are found in the general healthy population; and e) because many of them appear over time, appearing normal at birth. However, it is now known that some of these syndromes are caused by changes due to the loss of chromosome portions so small that they are not detected by high resolution cytogenetics (they are cryptic for these techniques), or to known mutations in certain genes.
As many of these children will present with their signs during childhood and are going to be seen by Primary Care doctors, it is essential that small guises are available to recognise them and manage them appropriately.
This article explains the different forms of presentation, their technical characteristics by which they can be diagnosed, as well as the symptoms that may make us aware of these syndromes and how to act. The prognosis very much depends on early identification and information to the family.
El nacimiento de un niño con defectos congénitos implica la necesidad de establecer un diagnóstico, aspecto que es muy importante por varios motivos. En primer lugar, porque «dar un nombre» al problema que presenta el recién nacido indica que ya se conoce y, por tanto, se puede tener información sobre las implicaciones que suponen el conjunto de sus defectos. Además, posibilita establecer una atención médica adecuada y, sobre todo, anticipatoria. En segundo lugar, porque implica también saber las potenciales causas, lo que permite determinar el riesgo de repetición familiar o su inexistencia. En tercer lugar, porque se podrá ofrecer una completa información a la familia, lo que disminuirá la angustia que suelen tener los padres cuando no saben lo que tiene su hijo.
Sin embargo, no siempre es posible establecer el diagnóstico de un recién nacido con defectos congénitos. Bien porque el conjunto de manifestaciones clínicas que presentan son aún de causa desconocida (en alrededor de un 55–60% de los casos) o porque los síndromes que ya se conocen tienen una frecuencia tan baja (la mayoría menores de 1 por cada 100.000 nacimientos, y en los más frecuentes de 2–3 por 100.000) que no son fáciles de reconocer cuando no se tiene experiencia previa; y mucho menos hacer el diagnóstico diferencial1. Además, forman cuadros sindrómicos que, aunque sean clínicamente similares, pueden ser causados por agentes o factores muy diferentes, y viceversa, síndromes clínicos muy distintos que se producen por alteraciones en un mismo gen.
Por todo eso, cuando nace un niño con defectos congénitos de cualquier tipo se deben seguir una serie de pasos para llegar a un diagnóstico.
¿Cuál es el sistema a seguir para ese diagnóstico?Lo primero es realizar una exploración clínica detallada, anotando no solo las alteraciones físicas, sino los percentiles de la somatometría y todos los rasgos dismórficos que presente cada paciente, independientemente de que algunos sean secundarios a otro defecto primario. Para esta exploración hay que realizar todas las pruebas que sean necesarias, tanto de imagen como de laboratorio. Además, e independientemente de que el conjunto de alteraciones sugiera un determinado síndrome mendeliano o ambiental, «siempre» se debe hacer un estudio citogenético con cromosomas de alta resolución (850 bandas). No se debe olvidar que la única alteración cromosómica que se puede «diagnosticar» por el fenotipo es la trisomía 21 (síndrome de Down) y que una alteración cromosómica puede presentar un amplio espectro de manifestaciones clínicas. Una vez descartadas las alteraciones cromosómicas visibles con cariotipos de 850 bandas, en algunos casos con determinadas alteraciones del desarrollo, se harán análisis citogenéticos con técnicas de hibridación in situ con fluorescencia (FISH en inglés) y en su caso otras más sofisticadas (que se expusieron en el artículo anterior).
¿Qué tipos de alteraciones se detectan con el cariotipo de alta resolución?Con los cromosomas de alta resolución (850 bandas) que se mostraron en el capítulo anterior de este curso se detectan alteraciones estructurales muy pequeñas, como pérdidas y ganancias de material cromosómico y también translocaciones (intercambios entre diferentes cromosomas) que pueden ser, o no, balanceadas. El tamaño mínimo de las alteraciones que se detectan con los cromosomas de alta resolución se encuentra en la frontera de 4 megabases (Mb). Sin embargo, cuando se han estudiado muchos casos con este tipo de cromosomas de alta resolución y se tiene la experiencia de haber podido identificar muchas alteraciones (como ocurre en el laboratorio del grupo del ECEMC) se han llegado a identificar alteraciones de 3Mb.
Cuando las alteraciones cromosómicas son menores de 3 Mb, ¿cómo se pueden detectar?Cuando el cariotipo de alta resolución es normal, se pueden aplicar técnicas de FISH, pero estas tienen que ser dirigidas por la clínica que presente el paciente. Hoy día se han reconocido diferentes cuadros clínicos producidos por deleciones en determinadas regiones de algunos cromosomas que se denominan «síndromes de microdeleción» (porque suelen tener un tamaño menor de 3Mb), para los que existen sondas específicas comercializadas. Por tanto, el paso siguiente al estudio citogenético de alta resolución es analizar estos síndromes, siempre que la clínica del niño permita sospechar que puede tener alguno de ellos. No obstante, para esto se debe tener un buen conocimiento de los distintos tipos de síndromes con alteraciones cromosómicas crípticas.
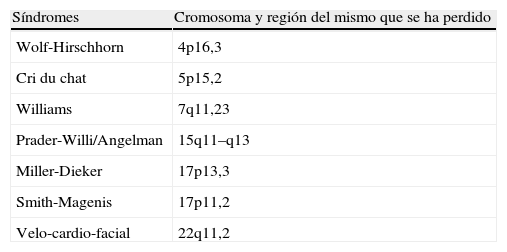
¿Cuáles son los más frecuentes y conocidos?Las microdeleciones más frecuentemente identificadas son las que dan lugar a los síndromes que se indican en la tabla 1. En los últimos tiempos se vienen caracterizando nuevos síndromes de microdeleción, aunque las características clínicas que inducen su sospecha solo se van delimitando cuando son muchos los casos clínicos que se han descrito con la microdeleción.
En realidad, la mayoría de los síndromes de microdeleción se consideran como «síndromes de gen contiguo», ya que algunas de sus manifestaciones clínicas se relacionan con el tamaño de la pérdida de ADN.
¿Cuáles serían las alteraciones más importantes para sospechar la microdeleción?Una vez que se ha concluido que el cariotipo de alta resolución (850 bandas) es normal, el camino a seguir debe venir determinado por las manifestaciones clínicas de cada paciente si se quiere hacer un uso adecuado de los recursos. Aunque no existe una relación patognomónica entre algún defecto y una sola causa (en este caso la microdeleción) en general, para los síndromes más frecuentes hasta la fecha (tabla 1 y fig. 1) se pueden seguir los siguientes pasos.
- 1.
Ante todo saber que, excepto el síndrome de la microdeleción 22q11.2 (síndrome velo-cardio-facial), cualquiera de los otros síndromes de la tabla 1 y la figura 1 van a presentar retraso mental de diferente gravedad.
- 2.
Si las manifestaciones clínicas del paciente incluyen retraso psicomotor y rasgos dismórficos u otros defectos congénitos más graves, pero no se reconoce síndrome alguno, se debe hacer un estudio de las regiones subteloméricas (generalmente mediante técnicas de MLPA)2.
- 3.
Cuando un recién nacido con diferentes defectos congénitos presente una configuración naso-frontal recta (que le da un aspecto clásicamente descrito como de casco de guerrero griego) se debe descartar el síndrome de Wolf-Hirschhorn (microdeleción 4p16.3)3,4.
- 4.
Si el recién nacido tiene, entre otros defectos, un llanto afónico y débil se debe descartar el síndrome de Cri du chat (microdeleción 5p15.2)5.
- 5.
Si el niño presenta estenosis aórtica supravalvular, habría que descartar que tuviera síndrome de Williams (microdeleción 7q11.23)6, aunque no se presenta en todos los casos.
- 6.
Si entre las manifestaciones clínicas incluyen hipotonía, se debe descartar la alteración correspondiente al síndrome de Prader-Willi/Angelman (microdeleción 15q11-q13). Sin embargo, este síndrome se puede producir también por otros mecanismos más sofisticados (disomía uniparental del cromosoma 15, o alteración de la metilación de la región crítica) que no se abordan en este curso7.
- 7.
Si tiene lisencefalia, habría que descartar, entre otros, el síndrome de Miller-Dieker (microdeleción 17p13.3)8,9.
- 8.
Si el niño presenta un comportamiento autolesivo y trastornos del sueño además de otros defectos, se debe pensar en el síndrome de Smith-Magenis (microdeleción 17p11.2)10.
- 9.
Si el niño presenta anomalías faciales, fisura del paladar o voz hipernasal y/o cardiopatía (incluso sin otras alteraciones), en un amplio espectro de manifestaciones clínicas, se debe descartar la microdeleción 22q11.211–13.

Aunque muchos de los defectos que permiten sospechar estos síndromes se presentan al nacimiento, dado que la intensidad de la manifestación puede variar mucho en algunos, junto con que el cuadro clínico general también es variable, en algunas ocasiones no se reconocen. Por ello, pueden ser niños que tarden en ser diagnosticados, y el que puedan ser identificados en Atención Primaria (AP) lo antes posible supondrá un indudable beneficio para ellos.
¿Cuándo se deben utilizar los CGH array?Aunque aún existen dificultades para interpretar los resultados de los CGH array en pacientes con defectos congénitos, dada su alta resolución cada vez se utilizan más en la práctica clínica. De hecho, algunos autores ya han empezado a considerar que en muy poco tiempo sustituirán a los cariotipos. Sin embargo, por ahora hay aspectos en contra de esta sustitución, por lo que es importante destacarlos. En primer lugar, el CGH array no es útil para detectar alteraciones estructurales balanceadas, por lo que debe ser un estudio complementario al cariotipo de alta resolución. En segundo lugar, para su interpretación es necesario tener una importante precisión de todas las alteraciones clínicas de cada paciente; entre otros aspectos, para interpretar los resultados del CGH, y su correlación con los defectos del paciente. Tercero, se necesita disponer de los datos resultantes del análisis con CGH array de una población sin defectos congénitos para interpretar correctamente si las variaciones observadas son, o no son, polimorfismos. Por último, son todavía técnicas muy caras para su utilización en todo niño con defectos congénitos o a modo de screening.
¿Y si el niño nace sin rasgos clínicos patológicos y luego evoluciona mal?En numerosas ocasiones, los recién nacidos que no muestran graves defectos congénitos, aunque tengan rasgos dismórficos, no son detectados al nacimiento. Además, dado que muchas alteraciones físicas del desarrollo embrionario (incluso no muy graves) son fáciles de identificar por diagnóstico prenatal, en muchos de los fetos afectados se va a decidir la interrupción voluntaria de la gestación. Esto tendrá un impacto sobre la frecuencia al nacimiento de las alteraciones físicas (que se expondrá en el siguiente capítulo de este curso).
Si las alteraciones del recién nacido no son físicas y su manifestación es evolutiva, solo se detectará que el niño tiene un problema cuando empiezan a presentarse durante los primeros años de vida. Esto puede producir los efectos siguientes:
- a.
Que los síndromes cuyas manifestaciones físicas sean muy leves y que presentan alteraciones psíquicas, bioquímicas o sensoriales, se van a manifestar durante los primeros años de vida. Por ello, son niños que generalmente se identificarán en AP.
- b.
Que al manifestarse durante la infancia, no va a ser fácil realizar un diagnóstico, ya que serán atendidos según se presenten las distintas alteraciones, por lo que con frecuencia van a pasar de médico en médico en busca de un diagnóstico que determine una atención integral a sus problemas.
- c.
Que no van a beneficiarse del tratamiento anticipatorio de sus potenciales manifestaciones, al no disponer de un diagnóstico.
- d.
La situación descrita en los apartados b y c va a producir una gran angustia en los padres y además, en muchos casos sobreexposición del niño a múltiples análisis que, en numerosas ocasiones, no están indicados.
Muchos de los aspectos anteriores pueden solucionarse si los médicos de atención primaria disponen de unos determinados conocimientos y unas guías para la actuación diagnóstica anticipatoria.
Guías anticipatorias para el diagnóstico de estos casos en AP- 1.
Cuando llega al médico de AP un niño con un síndrome de defectos congénitos ya diagnosticado:
En este caso, es muy importante que se pueda establecer un «diálogo» entre el médico de AP y el especialista que lleve el control del niño. En general, el médico de AP debería pedir a los padres que le lleven los informes que tengan junto con el tratamiento que han puesto al niño dependiendo de sus problemas y el plan de seguimiento. De esta forma, podrá conocer la evolución potencial y sus posibles complicaciones. Además, si el niño presenta un problema inesperado, la mejor actuación sería hablar con el especialista que lo está siguiendo; pero si esto no fuera posible, atender sintomáticamente el problema y derivar al niño al especialista que lleva su control. Entre las pruebas que deberían haberse hecho, deben estar las que se indican en el apartado siguiente, según la edad y datos clínicos y «siempre» un cariotipo de alta resolución aunque se hubiera diagnosticado un determinado síndrome.
- 2.
Cuando el problema no se detectó al nacimiento, es el médico de AP el que puede favorecer un diagnóstico precoz:
Como se indica en la tabla 2, en esta situación hay una serie de signos y síntomas que se pueden manifestar durante los primeros meses de vida o durante los primeros años que deben alertar al médico de AP. Así, ante cualquiera de las situaciones contempladas en la tabla 2 e independientemente de que envíe al niño al especialista que necesite para su control, el médico de AP debería hacer una detallada anamnesis y preguntar a los padres las pruebas y análisis que le han hecho. Si entre ellas no se incluyen las que se especifican seguidamente, el médico de AP debería hacer un informe con sus sospechas y enviarlo con los padres al servicio de genética del hospital que le corresponda. En dicho informe debería solicitar que cuando tengan los resultados les entreguen a los padres el informe para que ellos le entreguen una copia al médico de AP.
Tabla 2.Guías diagnósticas anticipatorias
Edad Problemas Primeros meses de vida Cuando los padres comentan que el bebé presenta Alteración de los estados de vigilia y sueño Movimientos oculares raros Llanto desconsolado durante periodos largos Vómitos frecuentes Posturas anormales como rigidez de tronco, cabeza, extremidades Que está siempre muy relajado y con poco tono en extremidades y postura como «flácida» Déficits sensoriales Que el bebé no sigue la pauta normal de adquisición para mantener la cabeza, sentarse, coger objetos… Durante los primeros años Tanto observado por los padres como en la exploración clínica en Atención Primaria Retraso del crecimiento (estatura, peso, perímetro cefálico) Retraso mental de cualquier grado, desde límite a muy grave Retraso del lenguaje Retraso de la marcha y/o de habilidades motoras Movimientos repetitivos Déficits sensoriales Obesidad Hiperfagia Agresividad y/o mala relación con otros niños Auto-agresiones, rabietas Falta de atención - a.
Cuando se trata de un recién nacido, o lactante (primera parte de latabla 2).
- i.
Si el niño tiene un problema neurológico y/o de retraso, incluyendo el de medro y psicomotor, y/o alguno de los problemas descritos en la primera parte de la tabla 2, se debería hacer un estudio cromosómico de alta resolución (850 bandas).
- ii.
Si el cariotipo de 850 bandas es normal, habría que hacer un estudio de las regiones subteloméricas de todos los cromosomas.
- iii.
Los dos estudios anteriores también están indicados si el niño presenta problemas que afectan a diversos órganos o sistemas.
- iv.
Si esos estudios fueran normales, dependiendo de la clínica, habría que descartar que tuviera alguno de los síndromes de microdelección.
- i.
- b.
Cuando se trate de un niño (segunda parte de latabla 2)
- i.
Enviar al niño al especialista que debe hacerle las pruebas correspondientes. No solo para determinar las causas físicas de las alteraciones (digestivas, neurológicas, cardiovasculares…), sino también bioquímicas y metabólicas. Pero también, indicando la necesidad de que se derive a un servicio de genética, para que le hagan los análisis anteriores, y puedan proporcionarles el asesoramiento genético, si es posible, junto con las posibilidades de detección en futuros embarazos.
- i.
Los autores declaran no tener ningún conflicto de intereses.
