







INTRODUCCIÓN
La esclerosis lateral amiotrófica (ELA) es la tercera forma más frecuente de enfermedad neurodegenerativa tras el Parkinson y la enfermedad de Alzheimer. Se caracteriza por la denervación de motoneuronas superiores e inferiores y tiene un curso progresivo y devastador.
Fue descrita por primera vez en 1874 por Charcot, pero hasta hace una década no se ha impulsado la investigación de sus mecanismos patogénicos y de tratamientos eficaces.
Aunque su pronóstico es fatal, el diagnóstico temprano permite descartar los casos potencialmente tratables que son confundidos con la ELA. Esta no es una tarea sencilla debido al comienzo insidioso de la enfermedad y al amplio abanico de síntomas con los que puede presentarse, siendo los más frecuentes la debilidad muscular progresiva en extremidades superiores, calambres, disfagia y disartria.
A continuación presentamos un caso en el que la observación de atrofia muscular de las manos de un paciente, que consultaba por otro motivo, permitió iniciar el estudio diagnóstico.
EXPOSICIÓN DEL CASO
Acude a la consulta de Atención Primaria un varón de 66 años por un episodio de lumbalgia de una semana de evolución. Indicamos al paciente que se descubra para realizar la exploración física de columna lumbar y observamos que presenta una gran dificultad para desabotonarse la
camisa así como para realizar otros movimientos finos y llama la atención una intensa atrofia en la musculatura de la mano en todos los territorios (figs. 1 y 2).
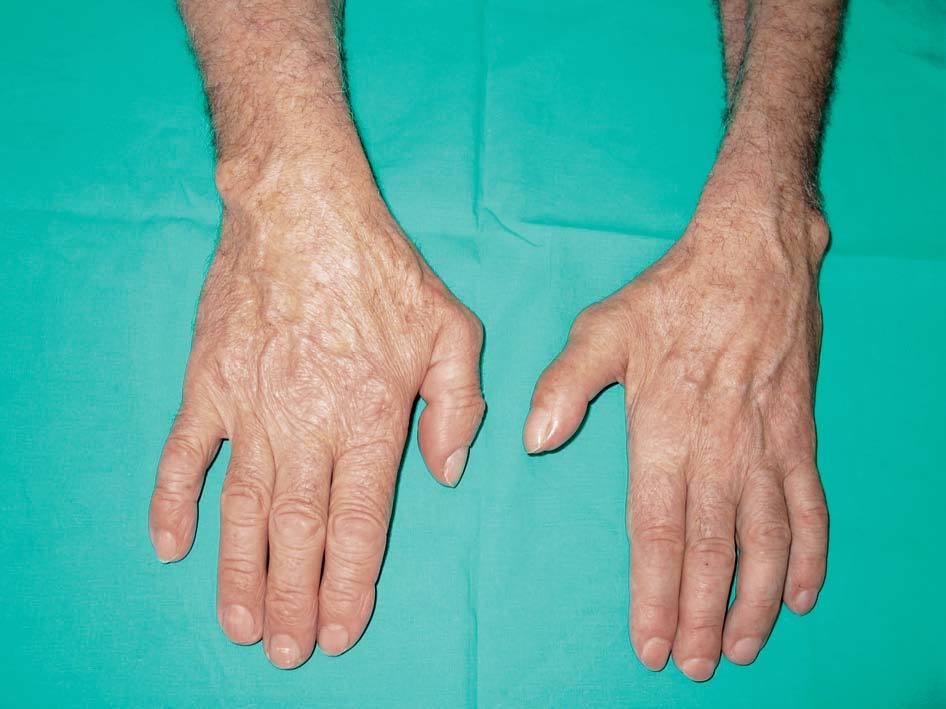
Figura 1. Cara dorsal de las manos.
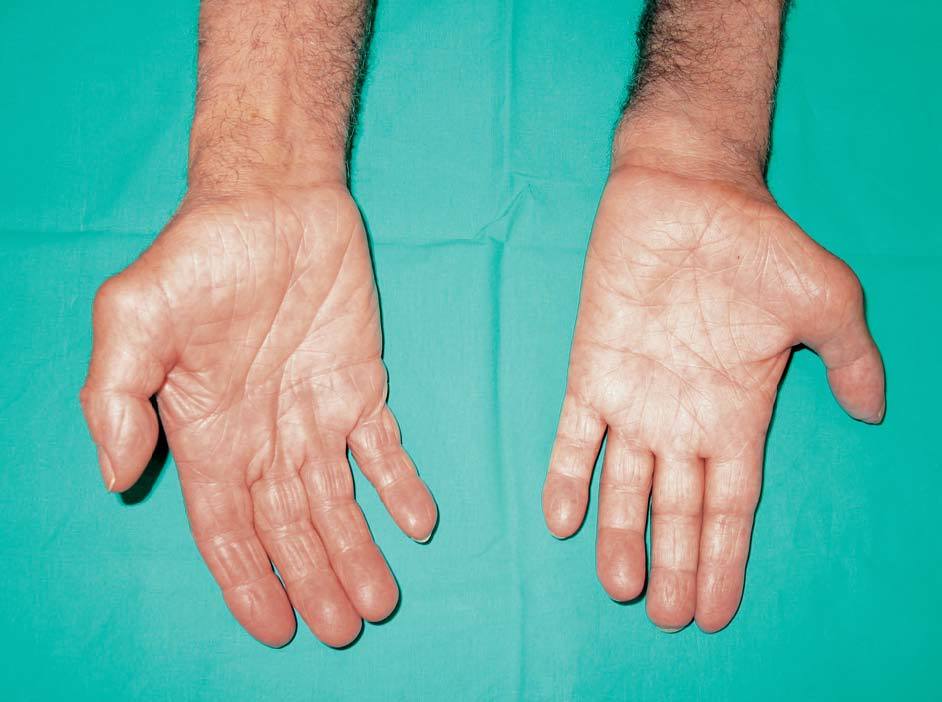
Figura 2. Cara palmar de las manos.
En la exploración física el paciente no presenta apofisalgia ni signos de elongación radicular, dolor a la flexoextensión de columna dorsolumbar, sin presentar limitación en los arcos de movimiento. La presión arterial (PA) es de 130/80 mmHg, sin hallazgos en la auscultación cardiopulmonar. En el examen neurológico no se observan alteraciones cognitivas y destaca una pérdida de fuerza en ambas extremidades superiores, de predominio izquierdo, sin presentar alteraciones de la sensibilidad, de la marcha ni signos cerebelosos. No se observan fasciculaciones, y los signos de Hoffmann, Babinsky, Tinnel y Phalen son negativos.
El paciente refiere que desde hace año y medio presenta pérdida de fuerza progresiva en ambas manos más marcada en la izquierda, que en los últimos meses ha evolucionado con mayor rapidez. Se ha visto obligado a jubilarse anticipadamente, al no poder realizar los movimientos minuciosos que precisa para su trabajo de electricista, aunque nunca ha consultado por ello.
Se diagnostica de lumbalgia aguda mecánica y ante el cuadro de atrofia muscular de las manos nos planteamos el siguiente diagnóstico diferencial (tabla 1).

Solicitamos hemograma, bioquímica, pruebas reumáticas, proteinograma, radiografías cervical y lumbar, electromiograma (EMG) e interconsulta con Neurología. Los parámetros de la analítica sanguínea estaban en rangos normales, las radiografías mostraban signos degenerativos compatibles con artrosis y en el EMG se observa degeneración en extensor común de los dedos, palmar mayor, primer interóseo dorsal, abductor del quinto dedo y tenar izquierdos. No se observan fasciculaciones ni signos de atrapamiento nervioso y la velocidad de conducción tanto motora como sensitiva son normales.
Seis meses después el paciente presenta un evidente empeoramiento de la amiotrofia en manos y antebrazos y se observa leve atrofia de músculo cuádriceps derecho.
Se realiza resonancia magnética nuclear (RMN) con compresiones discales de C5-C7 que comprimen el diámetro sagital del canal. En el último EMG se observa abundante actividad espontánea de denervación (fasciculaciones y ondas positivas) así como datos de denervación subcrónica segmentaria. El paciente es diagnosticado de ELA, comenzando tratamiento con riluzol.
DISCUSIÓN
Se detectan entre 1,2-1,8 casos por cada 100.000 habitantes al año1 de ELA y su prevalencia está estimada entre 4-6 casos cada 100.000 habitantes. Esta patología aumenta con la edad1,2 y es algo más frecuente en el sexo masculino (entre 1,3 y 1,6:1 hombre: mujer)3. La edad media de comienzo son los 58 años4.
Podemos distinguir tres formas:
1) Esporádica: 90-95% de los casos3.
2) Familiar (5-10% de los casos)3: por mutaciones halladas en el gen de la enzima superóxido dismutasa Cu-Zn dependiente (SOD 1) de herencia autosómico dominante.
3) Forma del Pacífico Occidental: en las regiones de Guam, Nueva Guinea Papúa y la isla de Kii, que constituyen un foco endémico con una prevalencia 50 veces mayor5 a la media en una variedad que asocia ELA con demencia y enfermedad de Parkinson.
Actualmente se barajan 6 hipótesis etiológicas que intentan explicar el origen de las formas esporádicas:
1) Excitotóxica6: el cúmulo de determinados neurotransmisores, como el glutamato, puede ser el origen de la muerte de las neuronas motoras.
2) Estrés oxidativo6: por cúmulo de radicales libres en las motoneuronas.
3) Inmunológica3,7: por mecanismos de autoinmunidad, síndromes paraneoplásicos, basadas en la presencia de anticuerpos antigangliósidos en algunos pacientes.
4) Trófica8: defiende que el déficit de factores tróficos transportados por flujo axonal retrógrado causa muerte neuronal.
5) Infecciosa6: relaciona la enfermedad con infección por virus de la familia enterovirus, retrovirus o priones.
6) Exposición a tóxicos7: fundamentalmente metales pesados y pesticidas.
El cuadro clínico surge de la combinación de signos y síntomas del compromiso de las neuronas motoras superiores (MNS) e inferiores (MNI). Las manifestaciones varían en función de si las motoneuronas más afectadas son las corticoespinales o las inferiores del tronco encefálico y médula espinal, aunque independientemente del predominio de la afectación inicial, la ELA presenta una progresión inexorable en la que se acaban afectando ambos tipos.
La denervación de las MNI de médula espinal cursa con debilidad muscular segmentaria (suele comenzar en zonas distales de las extremidades superiores), atrofia muscular, hiporreflexia, fasciculaciones y calambres musculares. Si se afectan las MNI bulbares presenta disartria, disfagia, hipotonía y fasciculaciones en la lengua.
Si son las MNS (corticoespinales) las que están dañadas aparece hiperreflexia, clonus, espasticidad, Babinsky, signo de Hoffmann y síntomas pseudobulbares.
Permanecen conservadas las funciones sensitiva y cognitiva y no existe compromiso de esfínteres ni de la musculatura ocular extrínseca.
El diagnóstico de la ELA es fundamentalmente clínico al no existir ninguna prueba específica. Sólo el examen electromiográfico tiene un valor equivalente a la clínica.
Según los criterios diagnósticos establecidos por la Federación Mundial de Neurología se requiere la presencia de9:
1) Signos de compromiso de la MNI en al menos una de las regiones bulbar, cervical, torácica o lumbosacra, por examen clínico, electrofisiológico o neuropatológico.
2) Signos de compromiso de la MNS en al menos una de las regiones anteriormente citadas por examen clínico.
3) Progresión de estos signos a más regiones.
También se requiere la ausencia de evidencias electrobiológicas o de neuroimagen de otras patologías que puedan explicar los signos de denervación de MNS y/o MNI10.
Se han establecido 4 niveles de certeza diagnóstica (ELA definida, probable, posible y sospechosa) en función de la combinación de los signos en las diferentes regiones anteriormente citadas11.
El curso de la enfermedad es inexorablemente progresivo, llevando al paciente a una inmovilidad casi completa con severos trastornos deglutorios y respiratorios por debilidad de la musculatura intercostal y diafragmática, causa más frecuente de muerte en estos pacientes. Su pronóstico es fatal, el 50% de los pacientes fallecen antes de los 4 años desde el comienzo de los síntomas5,7,12, un 20% y 5% tienen una supervivencia de 5 y 10 años respectivamente.
Dado que actualmente la ELA carece de tratamiento eficaz, es importante hacer especial hincapié en diferenciarla de todos aquellos casos potencialmente tratables; radiculopatías, neuropatía por atrapamiento, secundarios a patología endocrina y especialmente con la neuropatía motora multifocal (que responde al tratamiento con inmunoglobulinas por vía intravenosa o ciclofosfamida) con la que es frecuentemente confundida.
El único fármaco aprobado para su tratamiento es el riluzol (inhibidor de la liberación de glutamato), que produce una discreta prolongación de la supervivencia de entre 3 y 6 meses13,14. Por este motivo se debe poner especial énfasis en las medidas paliativas y rehabilitadoras adecuando los recursos a los problemas clínicos, predecibles en su mayoría, que van surgiendo, evitando el abandono que se hace de los pacientes con ELA al confundir enfermedad incurable con intratable.
