







Este documento de posicionamiento, auspiciado por la Asociación Española de Gastroenterología, la Sociedad Española de Oncología Médica, la Asociación Española de Genética Humana y el consorcio IMPaCT-Genómica, tiene como objetivo realizar recomendaciones para el uso de paneles de genes en la evaluación de individuos con alto riesgo de cáncer digestivo hereditario. Para medir la calidad de la evidencia y los niveles de recomendación se ha utilizado la metodología basada en el sistema Grading of Recommendations Assessment, Development and Evaluation (GRADE). Se obtuvo el consenso entre expertos mediante un método Delphi. El documento incluye recomendaciones sobre escenarios clínicos en los que se recomienda el uso de paneles de genes en cáncer colorrectal, síndromes polipósicos, cáncer gástrico y pancreático, así como los genes de los paneles a ser considerados en cada una de estas situaciones clínicas. También se establecen recomendaciones sobre la evaluación de mosaicismos, las estrategias de asesoramiento ante la ausencia de sujeto índice y, finalmente, el análisis constitucional tras identificación de variantes patogénicas tumorales.
This position statement, sponsored by the Asociación Española de Gastroenterología, the Sociedad Española de Oncología Médica, the Asociación Española de Genética Humana and the IMPaCT-Genómica Consortium aims to establish recommendations for use of multi-gene panel testing in patients at high risk of hereditary gastrointestinal and pancreatic cancer. To rate the quality of the evidence and the levels of recommendation, we used the methodology based on the GRADE system (Grading of Recommendations Assessment, Development and Evaluation). We reached a consensus among experts using a Delphi method. The document includes recommendations on clinical scenarios where multi-gene panel testing is recommended in colorectal cancer, polyposis syndromes, gastric and pancreatic cancer, as well as the genes to be considered in each clinical scenario. Recommendations on the evaluation of mosaicisms, counseling strategies in the absence of an index subject and, finally, constitutional analysis after identification of pathogenic tumor variants are also made.
Los cánceres digestivos se asocian, con una frecuencia variable, a variantes genéticas constitucionales, y, por tanto, heredables. La detección de variantes patogénicas asociadas al desarrollo de tumores digestivos tiene una doble implicación: por una parte, puede tener un impacto tanto pronóstico como predictivo de respuesta al tratamiento del tumor, y por otro, puede ser de gran relevancia para establecer estrategias preventivas y terapéuticas, tanto para el propio individuo como para sus familiares. Todas estas implicaciones deben ser valoradas de forma individualizada dentro de un proceso de asesoramiento o consejo genético, incluyendo la recogida sistemática de la historia personal y familiar, la información pretest de los posibles resultados, la visión de la población, los riesgos futuros y las estrategias preventivas que se pueden recomendar tanto a él como a sus familiares.
En las últimas décadas, la introducción de la tecnología de secuenciación de última generación Next Generation Sequencing (NGS) ha revolucionado la identificación de las variantes patogénicas en el ADN, permitiendo simplificar y reducir los costes del análisis además de aumentar considerablemente la información obtenida en el mismo acto con respecto a las tecnologías precedentes. Esta tecnología también se ha introducido en la práctica habitual del análisis de sospecha de cáncer hereditario, mediante el uso de paneles de genes. El uso de estos paneles permite secuenciar simultáneamente varios genes potencialmente implicados en el fenotipo observado, pero también implica un reto en la interpretación de los resultados al multiplicar el número de variantes genéticas detectadas. Según las recomendaciones del Colegio Americano de Genética Médica (ACMG)1, las variantes constitucionales detectables en un análisis genético pueden clasificarse en 5 niveles: benigna (clase 1), probablemente benigna (clase 2), variante de significado clínico incierto o desconocido (VUS/VSD) (clase 3), variante probablemente patogénica (clase 4) y variante patogénica (clase 5). Las variantes benignas y probablemente benignas no tienen implicación en enfermedad y pueden ser muy frecuentes en la población general. A fines prácticos, las variantes probablemente patogénicas suelen tratarse como si fueran patogénicas. Para simplificar la lectura del texto, las variantes probablemente patogénicas se agruparán con las patogénicas en la redacción del documento. En el caso de las variantes VUS o VSD, se recomienda revisar periódicamente su clasificación por si aparece nueva información científica que permita reclasificarlas como patogénicas o benignas. Otro factor a tener en cuenta en el proceso de asesoramiento genético es que no todas las variantes genéticas conllevan el mismo riesgo de desarrollar enfermedad, ya que pueden ser de baja, moderada o alta penetrancia. Además, el análisis simultáneo de varios genes puede ofrecer, en ocasiones, hallazgos incidentales o secundarios, como la detección de variantes patogénicas no claramente relacionadas con la sospecha evaluada. Esto ocurre, por ejemplo, con el caso de variantes patogénicas en los genes BRCA1 o BRCA2 en pacientes con agregación familiar de cáncer colorrectal. En este sentido, se definen los hallazgos incidentales como los que pueden tener o no implicaciones potenciales para la salud y significado clínico, pero no están relacionados con los síntomas de la enfermedad por los que se solicitó la prueba (p. ej., individuo con sospecha de síndrome de Lynch en el que se secuenciase el exoma con el fin de analizar un panel virtual de cáncer hereditario y se detectase una variante patogénica en MYH7, no contenido en el panel virtual y asociado con miocardiopatía hipertrófica). En contraste, se consideran como secundarios aquellos intencionadamente buscados y secundarios al objetivo que motivó la secuenciación (p. ej., mismo individuo y mismo test que en el ejemplo anterior, pero en el que se detectase una variante patogénica en CDH1, contenido en el panel virtual, pero asociado con cáncer gástrico [CG] difuso y cáncer de mama lobulillar)2.
El objetivo de este documento es establecer recomendaciones sobre el uso de paneles de genes en la evaluación de síndromes hereditarios asociados al cáncer digestivo. Concretamente, nos proponemos:
- 1.
Definir los escenarios clínicos en los que está indicado el análisis genético para la identificación de síndromes hereditarios asociados a neoplasias digestivas.
- 2.
Determinar qué genes se recomienda, actualmente, evaluar en cada escenario clínico.
- 3.
Establecer las indicaciones de estudio genético en mucosa sana en casos de estudio genético no informativo, para despistaje de mosaicismos.
- 4.
Establecer las indicaciones de estudio genético en tejido sano o familiares sanos en caso de que el caso índice no esté disponible.
- 5.
Establecer las indicaciones de estudio genético constitucional a partir de variantes patogénicas detectadas en la secuenciación de tumores digestivos.
Quedan fuera del ámbito de este documento proponer estrategias ante situaciones inciertas tales como la evaluación de las variantes genéticas de significado clínico incierto, los genes de baja penetrancia y los genes no asociados a la sospecha de cáncer hereditario observada (disociación fenotipo-genotipo).
MetodologíaEste documento surge como una colaboración de la Asociación Española de Gastroenterología (AEG), la Sociedad Española de Oncología Médica (SEOM), la Asociación Española de Genética Humana (AEGH) y el consorcio IMPaCT-Genómica3 con el fin de aunar criterios y recomendaciones tanto para gastroenterólogos clínicos como para oncólogos, médicos de familia y asesores genéticos. Se ha constituido un grupo de trabajo formado por expertos de las 4 sociedades/consorcios. Este grupo de trabajo se ha encargado de proponer y determinar las preguntas a responder en cada escenario clínico, a llevar a cabo la revisión sistemática de la literatura y a sintetizar los resultados de la misma. El panel de expertos decidió utilizar la terminología panel de genes frente a otras opciones disponibles (panel multi-gen) y el término constitucional para describir las variantes genéticas hereditarias detectadas en lo que hasta ahora se había definido como línea germinal, término que ahora se restringe al análisis del epitelio germinal de gónadas o gametos. Para establecer los niveles de evidencia y grados de recomendación de las diferentes preguntas evaluadas se ha utilizado la metodología basada en el sistema Grading of Recommendations Assessment, Development and Evaluation Working Group (GRADE)4. El sistema GRADE se puede aplicar tanto a la evaluación del riesgo como al efecto de las intervenciones y contextos, y equilibra la sencillez con la necesidad de considerar de forma global y transparente todos los aspectos importantes para hacer una recomendación. Una vez realizadas las recomendaciones, hemos evaluado el acuerdo entre los miembros del grupo de trabajo. El consenso se obtuvo utilizando el método Delphi. Las recomendaciones fueron evaluadas por los miembros del panel mediante una escala Likert: 1) Completamente en desacuerdo; 2) En desacuerdo; 3) Dudoso o con reparos; 4) De acuerdo, y 5) Completamente de acuerdo. En caso de desacuerdo se reformuló y votó nuevamente la recomendación. Se presentan y fundamentan las que obtuvieron un nivel de acuerdo superior al 80%. Las recomendaciones propuestas (tanto las consensuadas, como las no consensuadas) se pueden consultar en la tabla 1.
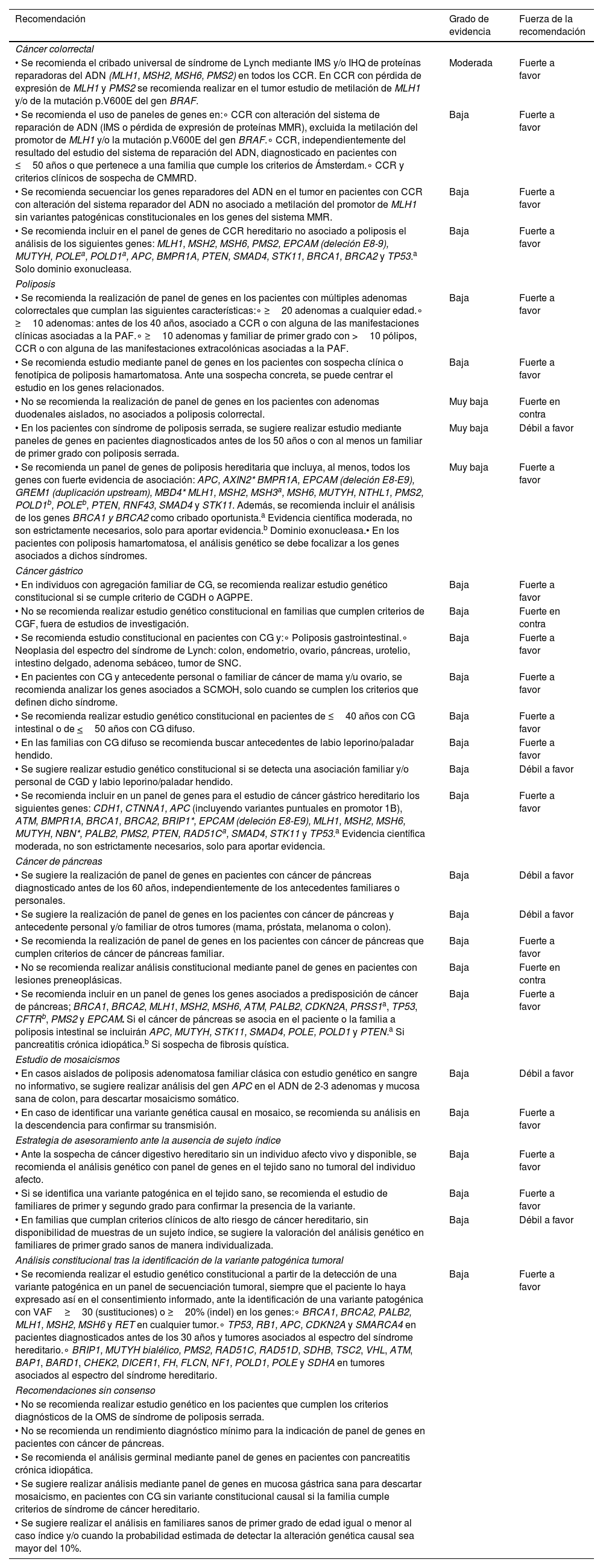
Recomendaciones del documento de posicionamiento sobre el uso de paneles de genes en la población de alto riesgo de cáncer digestivo hereditario
| Recomendación | Grado de evidencia | Fuerza de la recomendación |
|---|---|---|
| Cáncer colorrectal | ||
| • Se recomienda el cribado universal de síndrome de Lynch mediante IMS y/o IHQ de proteínas reparadoras del ADN (MLH1, MSH2, MSH6, PMS2) en todos los CCR. En CCR con pérdida de expresión de MLH1 y PMS2 se recomienda realizar en el tumor estudio de metilación de MLH1 y/o de la mutación p.V600E del gen BRAF. | Moderada | Fuerte a favor |
| • Se recomienda el uso de paneles de genes en:∘ CCR con alteración del sistema de reparación de ADN (IMS o pérdida de expresión de proteínas MMR), excluida la metilación del promotor de MLH1 y/o la mutación p.V600E del gen BRAF.∘ CCR, independientemente del resultado del estudio del sistema de reparación del ADN, diagnosticado en pacientes con ≤50 años o que pertenece a una familia que cumple los criterios de Ámsterdam.∘ CCR y criterios clínicos de sospecha de CMMRD. | Baja | Fuerte a favor |
| • Se recomienda secuenciar los genes reparadores del ADN en el tumor en pacientes con CCR con alteración del sistema reparador del ADN no asociado a metilación del promotor de MLH1 sin variantes patogénicas constitucionales en los genes del sistema MMR. | Baja | Fuerte a favor |
| • Se recomienda incluir en el panel de genes de CCR hereditario no asociado a poliposis el análisis de los siguientes genes: MLH1, MSH2, MSH6, PMS2, EPCAM (deleción E8-9), MUTYH, POLEa, POLD1a, APC, BMPR1A, PTEN, SMAD4, STK11, BRCA1, BRCA2 y TP53.a Solo dominio exonucleasa. | Baja | Fuerte a favor |
| Poliposis | ||
| • Se recomienda la realización de panel de genes en los pacientes con múltiples adenomas colorrectales que cumplan las siguientes características:∘ ≥20 adenomas a cualquier edad.∘ ≥10 adenomas: antes de los 40 años, asociado a CCR o con alguna de las manifestaciones clínicas asociadas a la PAF.∘ ≥10 adenomas y familiar de primer grado con >10 pólipos, CCR o con alguna de las manifestaciones extracolónicas asociadas a la PAF. | Baja | Fuerte a favor |
| • Se recomienda estudio mediante panel de genes en los pacientes con sospecha clínica o fenotípica de poliposis hamartomatosa. Ante una sospecha concreta, se puede centrar el estudio en los genes relacionados. | Baja | Fuerte a favor |
| • No se recomienda la realización de panel de genes en los pacientes con adenomas duodenales aislados, no asociados a poliposis colorrectal. | Muy baja | Fuerte en contra |
| • En los pacientes con síndrome de poliposis serrada, se sugiere realizar estudio mediante paneles de genes en pacientes diagnosticados antes de los 50 años o con al menos un familiar de primer grado con poliposis serrada. | Muy baja | Débil a favor |
| • Se recomienda un panel de genes de poliposis hereditaria que incluya, al menos, todos los genes con fuerte evidencia de asociación: APC, AXIN2* BMPR1A, EPCAM (deleción E8-E9), GREM1 (duplicación upstream), MBD4* MLH1, MSH2, MSH3a, MSH6, MUTYH, NTHL1, PMS2, POLD1b, POLEb, PTEN, RNF43, SMAD4 y STK11. Además, se recomienda incluir el análisis de los genes BRCA1 y BRCA2 como cribado oportunista.a Evidencia científica moderada, no son estrictamente necesarios, solo para aportar evidencia.b Dominio exonucleasa.• En los pacientes con poliposis hamartomatosa, el análisis genético se debe focalizar a los genes asociados a dichos síndromes. | Muy baja | Fuerte a favor |
| Cáncer gástrico | ||
| • En individuos con agregación familiar de CG, se recomienda realizar estudio genético constitucional si se cumple criterio de CGDH o AGPPE. | Baja | Fuerte a favor |
| • No se recomienda realizar estudio genético constitucional en familias que cumplen criterios de CGF, fuera de estudios de investigación. | Baja | Fuerte en contra |
| • Se recomienda estudio constitucional en pacientes con CG y:∘ Poliposis gastrointestinal.∘ Neoplasia del espectro del síndrome de Lynch: colon, endometrio, ovario, páncreas, urotelio, intestino delgado, adenoma sebáceo, tumor de SNC. | Baja | Fuerte a favor |
| • En pacientes con CG y antecedente personal o familiar de cáncer de mama y/u ovario, se recomienda analizar los genes asociados a SCMOH, solo cuando se cumplen los criterios que definen dicho síndrome. | Baja | Fuerte a favor |
| • Se recomienda realizar estudio genético constitucional en pacientes de ≤40 años con CG intestinal o de <50 años con CG difuso. | Baja | Fuerte a favor |
| • En las familias con CG difuso se recomienda buscar antecedentes de labio leporino/paladar hendido. | Baja | Fuerte a favor |
| • Se sugiere realizar estudio genético constitucional si se detecta una asociación familiar y/o personal de CGD y labio leporino/paladar hendido. | Baja | Débil a favor |
| • Se recomienda incluir en un panel de genes para el estudio de cáncer gástrico hereditario los siguientes genes: CDH1, CTNNA1, APC (incluyendo variantes puntuales en promotor 1B), ATM, BMPR1A, BRCA1, BRCA2, BRIP1*, EPCAM (deleción E8-E9), MLH1, MSH2, MSH6, MUTYH, NBN*, PALB2, PMS2, PTEN, RAD51Ca, SMAD4, STK11 y TP53.a Evidencia científica moderada, no son estrictamente necesarios, solo para aportar evidencia. | Baja | Fuerte a favor |
| Cáncer de páncreas | ||
| • Se sugiere la realización de panel de genes en pacientes con cáncer de páncreas diagnosticado antes de los 60 años, independientemente de los antecedentes familiares o personales. | Baja | Débil a favor |
| • Se sugiere la realización de panel de genes en los pacientes con cáncer de páncreas y antecedente personal y/o familiar de otros tumores (mama, próstata, melanoma o colon). | Baja | Débil a favor |
| • Se recomienda la realización de panel de genes en los pacientes con cáncer de páncreas que cumplen criterios de cáncer de páncreas familiar. | Baja | Fuerte a favor |
| • No se recomienda realizar análisis constitucional mediante panel de genes en pacientes con lesiones preneoplásicas. | Baja | Fuerte en contra |
| • Se recomienda incluir en un panel de genes los genes asociados a predisposición de cáncer de páncreas; BRCA1, BRCA2, MLH1, MSH2, MSH6, ATM, PALB2, CDKN2A, PRSS1a, TP53, CFTRb, PMS2 y EPCAM. Si el cáncer de páncreas se asocia en el paciente o la familia a poliposis intestinal se incluirán APC, MUTYH, STK11, SMAD4, POLE, POLD1 y PTEN.a Si pancreatitis crónica idiopática.b Si sospecha de fibrosis quística. | Baja | Fuerte a favor |
| Estudio de mosaicismos | ||
| • En casos aislados de poliposis adenomatosa familiar clásica con estudio genético en sangre no informativo, se sugiere realizar análisis del gen APC en el ADN de 2-3 adenomas y mucosa sana de colon, para descartar mosaicismo somático. | Baja | Débil a favor |
| • En caso de identificar una variante genética causal en mosaico, se recomienda su análisis en la descendencia para confirmar su transmisión. | Baja | Fuerte a favor |
| Estrategia de asesoramiento ante la ausencia de sujeto índice | ||
| • Ante la sospecha de cáncer digestivo hereditario sin un individuo afecto vivo y disponible, se recomienda el análisis genético con panel de genes en el tejido sano no tumoral del individuo afecto. | Baja | Fuerte a favor |
| • Si se identifica una variante patogénica en el tejido sano, se recomienda el estudio de familiares de primer y segundo grado para confirmar la presencia de la variante. | Baja | Fuerte a favor |
| • En familias que cumplan criterios clínicos de alto riesgo de cáncer hereditario, sin disponibilidad de muestras de un sujeto índice, se sugiere la valoración del análisis genético en familiares de primer grado sanos de manera individualizada. | Baja | Débil a favor |
| Análisis constitucional tras la identificación de la variante patogénica tumoral | ||
| • Se recomienda realizar el estudio genético constitucional a partir de la detección de una variante patogénica en un panel de secuenciación tumoral, siempre que el paciente lo haya expresado así en el consentimiento informado, ante la identificación de una variante patogénica con VAF≥30 (sustituciones) o ≥20% (indel) en los genes:∘ BRCA1, BRCA2, PALB2, MLH1, MSH2, MSH6 y RET en cualquier tumor.∘ TP53, RB1, APC, CDKN2A y SMARCA4 en pacientes diagnosticados antes de los 30 años y tumores asociados al espectro del síndrome hereditario.∘ BRIP1, MUTYH bialélico, PMS2, RAD51C, RAD51D, SDHB, TSC2, VHL, ATM, BAP1, BARD1, CHEK2, DICER1, FH, FLCN, NF1, POLD1, POLE y SDHA en tumores asociados al espectro del síndrome hereditario. | Baja | Fuerte a favor |
| Recomendaciones sin consenso | ||
| • No se recomienda realizar estudio genético en los pacientes que cumplen los criterios diagnósticos de la OMS de síndrome de poliposis serrada. | ||
| • No se recomienda un rendimiento diagnóstico mínimo para la indicación de panel de genes en pacientes con cáncer de páncreas. | ||
| • Se recomienda el análisis germinal mediante panel de genes en pacientes con pancreatitis crónica idiopática. | ||
| • Se sugiere realizar análisis mediante panel de genes en mucosa gástrica sana para descartar mosaicismo, en pacientes con CG sin variante constitucional causal si la familia cumple criterios de síndrome de cáncer hereditario. | ||
| • Se sugiere realizar el análisis en familiares sanos de primer grado de edad igual o menor al caso índice y/o cuando la probabilidad estimada de detectar la alteración genética causal sea mayor del 10%. | ||
CCR: cáncer colorrectal; CG: cáncer gástrico.
La prevalencia de síndromes hereditarios en pacientes con cáncer colorrectal (CCR) es de alrededor del 5-10%5–7. Clásicamente, el CCR hereditario se ha dividido, según el fenotipo, en asociado y no asociado a poliposis, en función de si aparece en contexto de múltiples pólipos colorrectales, generalmente 10 o más. En la predisposición hereditaria al CCR existe heterogeneidad genética (alteraciones en genes diferentes causan el mismo fenotipo clínico), expresividad variable (alteraciones genéticas en el mismo gen presentan diferentes fenotipos clínicos) y penetrancia incompleta (no todos los individuos con una misma variante genética manifiestan el fenotipo clínico asociado a ella)8. Esto, junto con la complejidad de la estrategia diagnóstica actual basada en la observación del fenotipo, y la creciente disponibilidad de paneles de genes, ha abierto la discusión de si plantear un estudio genético constitucional con paneles de genes a todos los pacientes con CCR con independencia de su fenotipo e historia familiar9. Sin embargo, hasta que lleguemos a ese punto, actualmente todavía se contempla una estrategia diagnóstica diferenciada para el fenotipo asociado y no asociado a poliposis.
Causas genéticas del cáncer colorrectal hereditarioLa principal causa del CCR hereditario no asociado a poliposis es el síndrome de Lynch (OMIM #120435, ORPHA:144), causado por la presencia de variantes patogénicas constitucionales monoalélicas en los genes de reparación de errores simples de apareamiento del ADN (sistema de reparación MisMatch Repair [MMR], genes: MLH1, MSH2, MSH6, PMS2), y las deleciones en la región 3’ del gen EPCAM, causantes de inactivación epigenética de MSH2. Este síndrome representa alrededor del 3% de todos los CCR10,11. Más del 95% de los tumores del síndrome de Lynch presentan alteración del sistema de reparación del ADN, definido como la presencia de inestabilidad de microsatélites (IMS) y/o pérdida de expresión de las proteínas reparadoras del ADN del sistema MMR, detectable por inmunohistoquímica (IHQ). Por otro lado, las alteraciones constitucionales bialélicas en los genes MMR conducen al síndrome de Deficiencia Constitucional de Reparación de Errores de Apareamiento (Constitutional MisMatch Repair Deficiency o CMMRD, OMIM #276300, ORPHA 252202), un síndrome minoritario caracterizado por el desarrollo de adenomas y CCR, tumores cerebrales y cáncer hematológico, principalmente en niños y adolescentes (tabla 2)12. La alteración del sistema MMR en tumores no es específica del síndrome de Lynch o CMMRD, y puede ocurrir por otras causas que inactivan los genes reparadores de apareamientos erróneos del ADN. De hecho, los síndromes de predisposición hereditaria son la causa menos frecuente de tumores con alteración del sistema MMR de reparación del ADN,13 siendo las principales causas:
- •
Metilación somática del promotor del gen MLH1. Esta es la causa más frecuente de tumores con pérdida de expresión de MLH1. Característicamente, estos tumores se asocian a variantes patogénicas somáticas en el protooncogén BRAF (excepcionalmente presente en el CCR con síndrome de Lynch). La presencia de variantes patogénicas en el gen BRAF (p.V600E) o la hipermetilación del promotor de MLH1 permiten clasificar esta forma de CCR como probablemente esporádico. Cabe destacar que, raramente, el síndrome de Lynch está causado por epimutaciones constitucionales en MLH1, caracterizadas por la hipermetilación monoalélica del promotor de MLH1 en tejido no neoplásico, que conduce al silenciamiento transcripcional del alelo afectado14.
- •
Doble mutación somática de los genes MMR. La inactivación de los dos alelos de alguno de los genes MLH1, MSH2, MSH6 o PMS2 mediante mutación puntual o pérdidas alélicas a nivel somático es otra causa frecuente de los tumores con alteración del sistema de reparación del ADN15–24. La secuenciación de los genes MMR en el tumor permite la detección de estas alteraciones somáticas (ausentes en línea germinal). Además, en una pequeña proporción de casos, variantes patogénicas constitucionales bialélicas en MUTYH o monoalélicas en POLE y POLD1 se han asociado a tumores con alteración del sistema MMR; en estos casos, la mutación somática de los genes reparadores ocurre como un evento secundario25–28.
Criterios de sospecha de Deficiencia Constitucional de Reparación de Errores de Apareamiento (Constitutional MisMatch Repair Deficiency)12. Se indicará estudio genético en pacientes con cáncer que puntúen ≥3 puntos
| Neoplasias y preneoplasias: una es obligatoria; si más de una neoplasia o preneoplasia está presente, sumar los puntos: | |
| • Carcinoma del espectro de síndrome de Lyncha diagnosticado antes de los 25 años | 3 |
| • Múltiples adenomas intestinales antes de los 25 años en ausencia de variantes patogénicas en APC/MUTYH o un adenoma con displasia de alto grado antes de los 25 años | 3 |
| • Glioma grado III o IV (OMS) diagnosticado antes de los 25 años | 2 |
| • Linfoma no Hodgkin de células T o PNETs diagnosticado antes de los 18 años | 2 |
| • Cualquier otro tumor diagnosticado antes de los 18 años | 1 |
| Características adicionales: opcionales; si más de una característica adicional está presente, sumar los puntos: | |
| • Signos clínicos de neurofibromatosis de tipo 1 y/o manchas hiperpigmentadas y/o hipopigmentadas en la piel de Ø>1cm | 2 |
| • Diagnóstico de síndrome de Lynch en un familiar de primer o segundo grado | 2 |
| • Carcinoma del espectro de síndrome de Lyncha diagnosticado antes de los 60 años en un familiar de primer, segundo o tercer grado | 1 |
| • Hermano con carcinoma del espectro de síndrome de Lyncha, glioma de alto grado, PNETs o linfoma no Hodgkin | 2 |
| • Hermano con cualquier tipo de tumor en la infancia | 1 |
| • Múltiples pilomatricomas en el paciente | 2 |
| • Un pilomatricoma en el paciente | 1 |
| • Agenesia del cuerpo calloso o cavernoma no inducido por terapia en el paciente | 1 |
| • Padres consanguíneos | 1 |
| • Deficiencia/niveles reducidos de IgG2/4 o IgA | 1 |
PNETs: tumor neuroectodérmico primitivo supratentorial.
Existen otros genes implicados en la reparación del ADN que se han asociado al CCR hereditario no polipósico con un sistema MMR indemne. Pese a que el número de genes propuestos es elevado (p, ej., MRE11, BARD1, POT1, BUB1B, POLE2, BRF1, IL12RB1, PTPN12 y PTPRJ), tan solo RPS20 ha demostrado una asociación consistente en el CCR hereditario29,30. Sin embargo, su frecuencia es extremadamente baja y no se dispone de suficientes datos sobre su fenotipo y penetrancia para poder realizar recomendaciones preventivas.
Criterios clínicos y moleculares para el diagnóstico de síndrome de LynchEl diagnóstico del síndrome de Lynch es complejo, dada la heterogeneidad de sus manifestaciones, y requiere de la identificación de una variante patogénica constitucional en los genes MMR. Por este motivo se desarrollaron diferentes criterios clínicos para identificar a los individuos y familias con mayor probabilidad de tenerlo: primero los criterios de Ámsterdam en 1990, posteriormente modificados en 1999 para incluir otros tumores diferentes al CCR. Los criterios de Bethesda recogen casi todas las condiciones clínicas asociadas al síndrome de Lynch con una sensibilidad superior al 90% y son de utilidad para seleccionar aquellos pacientes en los que realizar una determinación de IHQ o IMS31. Sin embargo, hoy en día la estrategia que ha demostrado mayor sensibilidad para la identificación del síndrome es el cribado universal en todos los CCR mediante estudio del sistema MMR en el tumor10,11. Además permite identificar tumores con un perfil pronóstico diferente, cuyo tratamiento puede verse modificado32.
Por otro lado, estudios recientes muestran que hasta el 16% (7-16% en diferentes series) de pacientes con diagnóstico de CCR no seleccionados por edad, historia personal o familiar de cáncer ni criterios moleculares, presentan variantes patogénicas en genes de susceptibilidad al cáncer, siendo más de la mitad en genes de alta penetrancia (incluidos genes como APC, BRCA1, BRCA2 o PALB2)7,33.
El CCR de aparición precoz (definido como aquel que ocurre antes de los 50 años) merece especial atención. La prevalencia de formas hereditarias de predisposición al CCR en esta subpoblación es superior a la del diagnosticado después de los 50 años, con una prevalencia global del 13% (rango de 9-26% en función de los estudios)34. Por ello, el rendimiento diagnóstico de los paneles de genes es superior en este escenario en comparación al CCR de aparición más tardía. La guía de la Asociación Americana de Oncología y la Red Nacional Integral del Cáncer (NCCN) del 2022 recomienda el uso de paneles constitucionales de genes a todos los pacientes con CCR<50 años con independencia del estudio del sistema MMR de reparación del ADN en el tumor o la historia personal o familiar35. El National Genomic Test Directory del NHS recomienda el uso de paneles de genes a todos los casos de CCR≤40 años36.
Ventajas del uso de paneles de genes en el cáncer colorrectal hereditarioEn el contexto del CCR hereditario no asociado a poliposis, el uso de paneles de genes ofrece las siguientes ventajas:
- •
El análisis secuencial de los genes reparadores en función del resultado de la IHQ MMR se ha simplificado enormemente, pudiendo analizar en paralelo todos los genes del sistema MMR. Este hecho ahorra tiempo y dinero en la estrategia diagnóstica37. El uso de paneles de genes en pacientes con CCR no asociado a poliposis tiene un rendimiento diagnóstico para síndrome de Lynch igual o superior a los criterios clínicos o la estrategia universal con estudio del sistema MMR38, dado que hasta el 5-10% de los tumores del síndrome de Lynch pueden presentar un sistema de reparación del ADN indemne o un resultado equívoco9. El estudio constitucional directo permite diagnosticar estos casos que, pese a ser poco frecuentes, permanecerían sin diagnosticar con la estrategia basada en el estudio del sistema de reparación de apareamientos erróneos del ADN en el tumor.
- •
El uso de un panel de genes permite incrementar el rendimiento diagnóstico de otras formas de CCR hereditario a parte del síndrome de Lynch. La inclusión en el panel de otros genes de alta penetrancia asociados al CCR (MUTYH, POLE, POLD1, APC, SMAD4, BMPR1A) permite diagnosticar formas hereditarias que podrían no sospecharse en base al fenotipo (expresividad variable)6,39,40. sí, las variantes patogénicas bialélicas en MUTYH predisponen a CCR no asociado a poliposis hasta en el 20% de los casos41 y se dispone de menos evidencia en cuanto al fenotipo de las variantes patogénicas en POLE y POLD142. También facilita la detección de otras formas de cáncer hereditario de alta penetrancia no sospechadas por criterios clínicos. Los estudios que han analizado paneles de genes en pacientes con CCR demuestran de forma consistente que en >BER1% de los casos se detectan variantes patogénicas constitucionales en genes como BRCA1, BRCA2, PALB2, CDKN2A o TP53, en los que la accionabilidad clínica ha sido demostrada (su detección se acompaña de medidas preventivas o terapéuticas específicas)6,7,39,43.
Dada la complejidad del algoritmo diagnóstico del síndrome de Lynch y la necesidad de realizar estudios somáticos (BRAF, metilación del ADN, secuenciación de MLH1, MSH2, MSH6, PMS2) y constitucionales (MLH1, MSH2, MSH6, PMS2, MUTYH, POLE, POLD1), se ha planteado la posibilidad de realizar un estudio de secuenciación en tumor de los genes reparadores del ADN inicial como estrategia de cribado, o incluso un estudio pareado simultáneo mediante secuenciación constitucional y somática en todos los casos de CCR9. La secuenciación del tumor permite identificar aquellos pacientes con síndrome de Lynch con una sensibilidad de 100% (IC 95%: 93,8-100%) y especificidad de 95,3% (IC 95%: 92,6-97,2%)9. Esta estrategia tendría la ventaja adicional de poder disponer de datos de secuenciación del tumor para personalizar terapias oncológicas. Sin embargo, el rendimiento y coste-efectividad de las estrategias de secuenciación universal todavía están por definir38.
Escenarios clínicos en los que se recomienda el uso de paneles de genes en el CCR no polipósico- •
Se recomienda el cribado universal de síndrome de Lynch mediante IMS y/o IHQ de proteínas reparadoras del ADN (MLH1, MSH2, MSH6, PMS2) en todos los CCR. En CCR con pérdida de expresión de MLH1 y PMS2 se recomienda realizar en el tumor estudio de metilación de MLH1 y/o de la mutación p.V600E del gen BRAF.
Calidad de la evidencia moderada, nivel de recomendación fuerte a favor.
- •
Se recomienda el uso de paneles de genes en:
- ∘
CCR con alteración del sistema de reparación de ADN (IMS o pérdida de expresión de proteínas MMR), excluida la metilación del promotor de MLH1 y/o la mutación p.V600E del gen BRAF.
- ∘
CCR, independientemente del resultado del estudio del sistema de reparación del ADN, diagnosticado en paciente con ≤BER50 años o que pertenece a una familia que cumple los criterios de Ámsterdam.
- ∘
CCR y criterios clínicos de sospecha de CMMRD.
- ∘
Calidad de la evidencia baja, nivel de recomendación fuerte a favor.
- •
Se recomienda secuenciar los genes reparadores del ADN en el tumor en pacientes con CCR con alteración del sistema reparador del ADN no asociado a metilación del promotor de MLH1 sin variantes patogénicas constitucionales en los genes del sistema MMR.
Calidad de la evidencia baja, nivel de recomendación fuerte a favor.
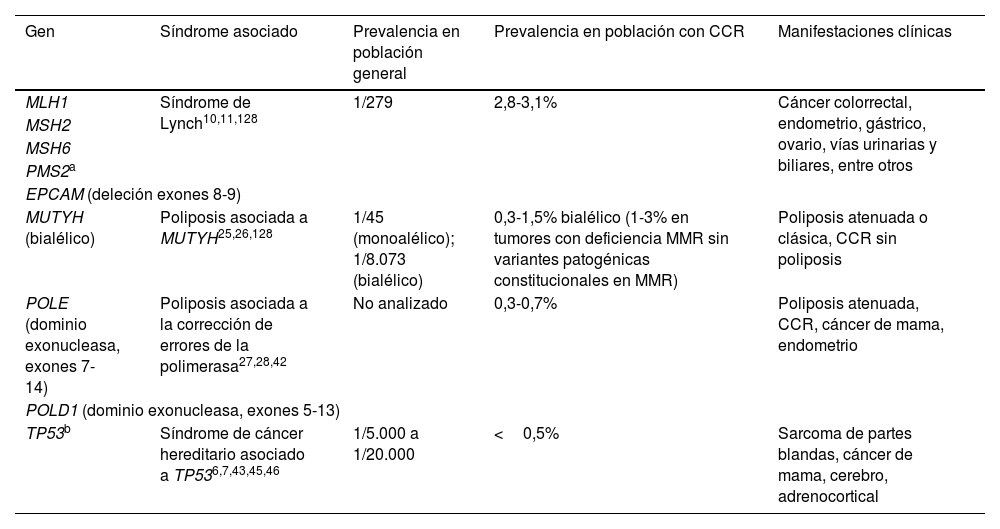
Genes a incluir en el panel de CCR no asociado a poliposisSe consideran genes relevantes para la evaluación genética del CCR hereditario los siguientes (tabla 3):
- •
Genes del sistema MMR responsables del síndrome de Lynch (MLH1, MSH2, MSH6, PMS2), incluyendo las deleciones en el extremo 3.’ de EPCAM (exones 8-9) que inducen el silenciamiento del gen contiguo MSH2. El análisis debe incluir el estudio de variantes puntuales y grandes reordenamientos. Debido a la presencia de pseudogenes altamente homólogos, el análisis del gen PMS2 se realizará si existe pérdida de expresión exclusiva de PMS2 en el tumor o en todos los casos de CCR, si se dispone de una técnica que haya demostrado validez analítica.
- •
El gen MUTYH, para el que variantes patogénicas constitucionales bialélicas se han asociado a tumores con variantes patogénicas somáticas en genes MMR25,26. El análisis debe incluir el estudio de variantes puntuales y grandes reordenamientos.
- •
Los genes POLE y POLD1, también asociados a variantes patogénicas somáticas en MMR. El análisis debe incluir el estudio de variantes puntuales en el dominio exonucleasa27,28.
- •
Además, se recomienda incluir el análisis de los genes BRCA1 y BRCA2 como cribado oportunista44 y TP53 en casos con CCR a edad joven (<50 años) o criterios de Chompret45,46.
- •
Asimismo, se define un grupo adicional de genes que representan una fracción global menor de CCR incidente pero que, no obstante, se asocian a un elevado riesgo de cáncer en los portadores:
Características principales de los genes relevantes para la evaluación genética del CCR hereditario
| Gen | Síndrome asociado | Prevalencia en población general | Prevalencia en población con CCR | Manifestaciones clínicas |
|---|---|---|---|---|
| MLH1 | Síndrome de Lynch10,11,128 | 1/279 | 2,8-3,1% | Cáncer colorrectal, endometrio, gástrico, ovario, vías urinarias y biliares, entre otros |
| MSH2 | ||||
| MSH6 | ||||
| PMS2a | ||||
| EPCAM (deleción exones 8-9) | ||||
| MUTYH (bialélico) | Poliposis asociada a MUTYH25,26,128 | 1/45 (monoalélico); 1/8.073 (bialélico) | 0,3-1,5% bialélico (1-3% en tumores con deficiencia MMR sin variantes patogénicas constitucionales en MMR) | Poliposis atenuada o clásica, CCR sin poliposis |
| POLE (dominio exonucleasa, exones 7-14) | Poliposis asociada a la corrección de errores de la polimerasa27,28,42 | No analizado | 0,3-0,7% | Poliposis atenuada, CCR, cáncer de mama, endometrio |
| POLD1 (dominio exonucleasa, exones 5-13) | ||||
| TP53b | Síndrome de cáncer hereditario asociado a TP536,7,43,45,46 | 1/5.000 a 1/20.000 | <0,5% | Sarcoma de partes blandas, cáncer de mama, cerebro, adrenocortical |
CCR: cáncer colorrectal.
- ∘
APC, asociado a poliposis adenomatosa familiar. La variante p.I1307K está asociada a un incremento de riesgo de CCR no asociado a poliposis en individuos con ascendencia Askhenazi, donde es especialmente prevalente47.
- ∘
BMPR1A y SMAD4, asociados a síndrome de poliposis juvenil, caracterizados por el desarrollo de pólipos hamartomatosos48. Se han reportado algunos casos de pacientes con CCR en ausencia de pólipos juveniles49.
- ∘
PTEN, asociado a síndrome de Cowden, caracterizado por el desarrollo de poliposis con múltiples histologías y mayor riesgo de CCR entre otros tumores50.
- ∘
STK11, asociado a síndrome de Peutz-Jeghers, que causa poliposis hamartomatosa y predisposición a varios tipos de tumores, entre ellos el CCR51.
Se recomienda incluir en el panel de genes de CCR hereditario no asociado a poliposis el análisis de los siguientes genes: MLH1, MSH2, MSH6, PMS2, EPCAM (deleción E8-9), MUTYH, POLEa, POLD1a, APC, BMPR1A, PTEN, SMAD4, STK11, BRCA1, BRCA2 y TP53.
aDominio exonucleasa.
Calidad de la evidencia baja, nivel de recomendación fuerte a favor.
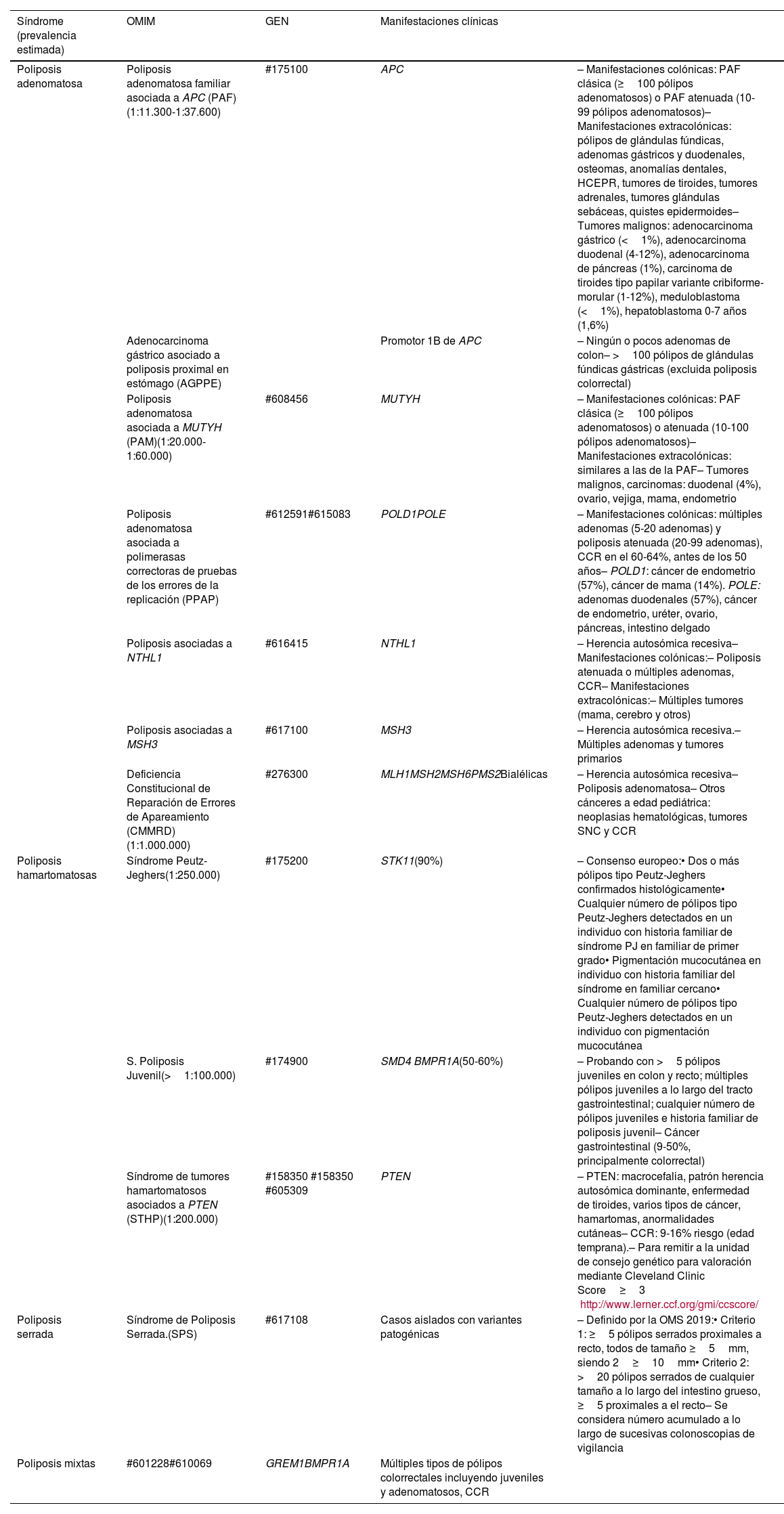
2. Uso de paneles de genes en las poliposis gastrointestinalIntroducción y revisión de la evidenciaLos síndromes de poliposis hereditaria se caracterizan por ser genética y clínicamente heterogéneos, con características clínicas que a menudo solapan en uno o más de los distintos síndromes. La identificación de la causa genética es fundamental para el pronóstico y seguimiento de los individuos afectos y portadores. Existen diferentes guías que establecen los criterios o indicaciones de estudio genético, según la histología y el número de pólipos, junto con otros factores. Estas indicaciones se basan en estudios retrospectivos o transversales, así como en opiniones de expertos52–55. Las poliposis colorrectales se clasifican, según el tipo histológico predominante, en poliposis adenomatosas, síndrome de poliposis serrada y poliposis hamartomatosas (histología predominante en >50% de los pólipos). Las recomendaciones generales y un resumen de estas pueden verse en la tabla 4.
Síndromes polipósicos asociados a cáncer colorrectal
| Síndrome (prevalencia estimada) | OMIM | GEN | Manifestaciones clínicas | |
|---|---|---|---|---|
| Poliposis adenomatosa | Poliposis adenomatosa familiar asociada a APC (PAF)(1:11.300-1:37.600) | #175100 | APC | – Manifestaciones colónicas: PAF clásica (≥100 pólipos adenomatosos) o PAF atenuada (10-99 pólipos adenomatosos)– Manifestaciones extracolónicas: pólipos de glándulas fúndicas, adenomas gástricos y duodenales, osteomas, anomalías dentales, HCEPR, tumores de tiroides, tumores adrenales, tumores glándulas sebáceas, quistes epidermoides– Tumores malignos: adenocarcinoma gástrico (<1%), adenocarcinoma duodenal (4-12%), adenocarcinoma de páncreas (1%), carcinoma de tiroides tipo papilar variante cribiforme-morular (1-12%), meduloblastoma (<1%), hepatoblastoma 0-7 años (1,6%) |
| Adenocarcinoma gástrico asociado a poliposis proximal en estómago (AGPPE) | Promotor 1B de APC | – Ningún o pocos adenomas de colon– >100 pólipos de glándulas fúndicas gástricas (excluida poliposis colorrectal) | ||
| Poliposis adenomatosa asociada a MUTYH (PAM)(1:20.000- 1:60.000) | #608456 | MUTYH | – Manifestaciones colónicas: PAF clásica (≥100 pólipos adenomatosos) o atenuada (10-100 pólipos adenomatosos)– Manifestaciones extracolónicas: similares a las de la PAF– Tumores malignos, carcinomas: duodenal (4%), ovario, vejiga, mama, endometrio | |
| Poliposis adenomatosa asociada a polimerasas correctoras de pruebas de los errores de la replicación (PPAP) | #612591#615083 | POLD1POLE | – Manifestaciones colónicas: múltiples adenomas (5-20 adenomas) y poliposis atenuada (20-99 adenomas), CCR en el 60-64%, antes de los 50 años– POLD1: cáncer de endometrio (57%), cáncer de mama (14%). POLE: adenomas duodenales (57%), cáncer de endometrio, uréter, ovario, páncreas, intestino delgado | |
| Poliposis asociadas a NTHL1 | #616415 | NTHL1 | – Herencia autosómica recesiva– Manifestaciones colónicas:– Poliposis atenuada o múltiples adenomas, CCR– Manifestaciones extracolónicas:– Múltiples tumores (mama, cerebro y otros) | |
| Poliposis asociadas a MSH3 | #617100 | MSH3 | – Herencia autosómica recesiva.– Múltiples adenomas y tumores primarios | |
| Deficiencia Constitucional de Reparación de Errores de Apareamiento (CMMRD)(1:1.000.000) | #276300 | MLH1MSH2MSH6PMS2Bialélicas | – Herencia autosómica recesiva– Poliposis adenomatosa– Otros cánceres a edad pediátrica: neoplasias hematológicas, tumores SNC y CCR | |
| Poliposis hamartomatosas | Síndrome Peutz-Jeghers(1:250.000) | #175200 | STK11(90%) | – Consenso europeo:• Dos o más pólipos tipo Peutz-Jeghers confirmados histológicamente• Cualquier número de pólipos tipo Peutz-Jeghers detectados en un individuo con historia familiar de síndrome PJ en familiar de primer grado• Pigmentación mucocutánea en individuo con historia familiar del síndrome en familiar cercano• Cualquier número de pólipos tipo Peutz-Jeghers detectados en un individuo con pigmentación mucocutánea |
| S. Poliposis Juvenil(>1:100.000) | #174900 | SMD4 BMPR1A(50-60%) | – Probando con >5 pólipos juveniles en colon y recto; múltiples pólipos juveniles a lo largo del tracto gastrointestinal; cualquier número de pólipos juveniles e historia familiar de poliposis juvenil– Cáncer gastrointestinal (9-50%, principalmente colorrectal) | |
| Síndrome de tumores hamartomatosos asociados a PTEN (STHP)(1:200.000) | #158350 #158350 #605309 | PTEN | – PTEN: macrocefalia, patrón herencia autosómica dominante, enfermedad de tiroides, varios tipos de cáncer, hamartomas, anormalidades cutáneas– CCR: 9-16% riesgo (edad temprana).– Para remitir a la unidad de consejo genético para valoración mediante Cleveland Clinic Score≥3 http://www.lerner.ccf.org/gmi/ccscore/ | |
| Poliposis serrada | Síndrome de Poliposis Serrada.(SPS) | #617108 | Casos aislados con variantes patogénicas | – Definido por la OMS 2019:• Criterio 1: ≥5 pólipos serrados proximales a recto, todos de tamaño ≥5mm, siendo 2≥10mm• Criterio 2: >20 pólipos serrados de cualquier tamaño a lo largo del intestino grueso, ≥5 proximales a el recto– Se considera número acumulado a lo largo de sucesivas colonoscopias de vigilancia |
| Poliposis mixtas | #601228#610069 | GREM1BMPR1A | Múltiples tipos de pólipos colorrectales incluyendo juveniles y adenomatosos, CCR |
CCR: cáncer colorrectal; HCEPR hipertrofia congénita del epitelio pigmentario de la retina.
Fuente: daptado de Guillén-Ponce C et al.53.
Poliposis adenomatosa familiar (PAF, OMIM#175100): Se trata de la poliposis hereditaria más frecuente. Presenta un patrón de herencia autosómico dominante y está causada por variantes constitucionales patogénicas en el gen APC (gen supresor tumoral que codifica una proteína que actúa como regulador negativo de la vía de transducción de señales Wnt. La gran mayoría de las variantes descritas en APC son de tipo truncante (frameshift o stopgain; 80-90%), o grandes deleciones (variantes de número de copia (CNV) que incluyen uno o más exones del gen (8-12%). El riesgo que tiene un individuo portador de transmitir la variante causal de APC a la descendencia es del 50%, y como es un síndrome de alta penetrancia, prácticamente el 100% de los individuos portadores tendrán alto riesgo a desarrollar PAF a lo largo de la vida.
Aproximadamente el 20% de los paciemtes con poliposis adenomatosa no presentan historia familiar de poliposis. Si estos pacientes son portadores de una variante constitucional en APC, esta pudo haber surgido ‘de novo’ en el paciemte, o heredarse de un progenitor con mosaicismo germinal. En caso de no identificarse ninguna alteración genética patogénica constitucional en estos pacientes debe contemplarse la posibilidad de que exista un mosaicismo somático. En estos casos, el riesgo de transmisión de la variante a la descendencia va a depender de si el mosaicismo afecta a las células germinales o está confinado en el colon (ver mosaicismos).
Poliposis asociada a MUTYH (PAM, OMIM#604933): Síndrome de herencia autosómica recesiva causado por variantes patogénicas bialélicas (homocigotas o heterocigotas compuestas) en el gen MUTYH. Este gen codifica una ADN-glicosilasa que participa en la reparación del daño oxidativo en el ADN (ruta de reparación por escisión de bases-BER). En población europea las dos variantes patogénicas recurrentes, c.536A>G, p.(Tyr179Cys) y c.1187G>A, p.(Gly396Asp) según NM_001128425.1, también denominadas c.452A>G, p.(Tyr151Cys) y c.1103G>A, p.(Gly368Asp) según NM_001048174.2 -MANE Select-, explicarían aproximadamente el 80% de las PAM. En MUTYH las variantes patogénicas descritas son mayoritariamente de tipo missense y alguna frameshift y, aunque las CNVs no son frecuentes, se ha descrito una gran deleción que incluye los exones 4 al 16 en población española, francesa y brasileña56. Los individuos portadores monoalélicos (heterocigotos) de variantes patogénicas en MUTYH presentan un riesgo moderado de desarrollar CCR, especialmente aquellos con un familiar de primer grado con CCR57. Se calcula que el 1% de la población europea son portadores monoalélicos de una de las 2 variantes recurrentes (bien de la p.Tyr179Cys o de la p.Gly396Asp)8,58.
Poliposis asociada a polimerasas correctoras de pruebas (PPAP; OMIM#612591; #615083): Síndrome de herencia autosómica dominante causado por variantes constitucionales patogénicas localizadas en el dominio exonucleasa de la polimerasa épsilon (POLE) y delta (POLD). Las variantes descritas son de tipo missense y se localizan en las regiones exónicas de POLD1 y POLE correspondientes a los dominios exonucleasa de las polimerasas. Los dominios exonucleasa les confieren a las polimerasas su capacidad correctora de pruebas (proofreading) para el mantenimiento de la fidelidad durante la replicación del ADN. Los individuos portadores pueden presentar poliposis clásica o atenuada, CCR y otros tumores hipermutados, generalmente con estabilidad de microsatélites (fenotipos proficient mismatch repair [pMMR]), como en el endometrio, aunque también se han descrito tumores inestables59.
Poliposis adenomatosa asociada a NTHL1 (PAN; OMIM#616415): Las variantes bialélicas patogénicas en NTHL1 son responsables de esta poliposis que se hereda de forma autosómica recesiva. NTHL1 codifica un ADN glicosilasa que participa en la ruta base-excision repair (VER), junto con MUTYH, en la reparación del ADN con daño oxidativo. Se ha descrito que las variantes bialélicas en este gen no solo incrementan el riesgo de poliposis y CCR, sino también de cáncer de mama60,61. A diferencia de MUTYH, los portadores monoalélicos de variantes patogénicas en NTHL1 no presentan un riesgo incrementado respecto a la población general de desarrollar tumores colorrectales u otros cánceres62.
Otros genes asociados a poliposis adenomatosas: Se han identificado variantes bialélicas en el gen MSH3, reparador de errores de apareamiento del ADN (MMR), en familias con poliposis y patrón de herencia autosómico recesivo. Como se ha mencionado previamente, las variantes bialélicas en los otros genes MMR son responsables del síndrome de CMMRD, que puede presentarse como múltiples pólipos, solapando con el fenotipo de otras poliposis con debut a edad joven12. Recientemente, se han identificado variantes bialélicas de pérdida de función en el gen MBD4, responsables de un síndrome recesivo caracterizado por múltiples adenomas colorrectales y neoplasias extracolónicas, como melanoma uveal y leucemias. MBD4 codifica una ADN-glicosilasa que forma parte del sistema de reparación BER63. El gen AXIN2 es responsable del síndrome de oligodoncia y CCR (OMIM# 604025, 608615), y la proteína que codifica participa en la degradación de la β-catenina en la ruta canónica de señalización Wnt.
Factores que predicen una causa hereditaria en pacientes con múltiples adenomasEn las poliposis adenomatosas se reconocen dos formas clínicas en función del número de adenomas colorrectales acumulados en una o varias colonoscopias: la forma clásica (≥100 adenomas) y la atenuada (<100)7,8. La probabilidad de encontrar una variante patogénica constitucional responsable del fenotipo se asocia al número de adenomas, la edad al diagnóstico y losantecedentes personales y/o familiares de CCR o de manifestaciones extracolónicas. En relación al número de adenomas, es habitual detectar la variante genética responsable del fenotipo en las formas clásicas (≥100 adenomas)64, mientras que solo en el 13-18% de los pacientes con 20-99 adenomas se detectan variantes patogénicas en alguno de los 2 genes más frecuentes (APC/MUTYH) y no llega al 5% en pacientes con <20 adenomas65,66. Por otro lado, el incremento en la edad de diagnóstico de la poliposis disminuye significativamente la probabilidad de detectar una variante genética responsable. En base al estudio de Stanich et al.65, con más de 3.000 individuos en la cohorte de múltiples adenomas, si se estableciera una probabilidad pre-test del 5% para detección de una causa hereditaria, estaría indicado el estudio genético con panel de genes en individuos con ≥10 adenomas antes de los 40 años o ≥20 adenomas antes de los 70.
Poliposis hamartomatosasSe caracterizan por la presencia de pólipos gastrointestinales de tipo hamartomatoso y manifestaciones extracolónicas (como cáncer de mama, páncreas, gástrico, etc.). Son síndromes raros de herencia autosómica dominante e incluyen:
Síndrome de Peutz-Jeghers (SPJ, OMIM#175200) causado por variantes patogénicas en STK11, que codifica una quinasa que forma parte de la ruta mTOR. Este síndrome se caracteriza por la presencia de manchas mucocutáneas hiperpigmentadas, poliposis gastrointestinal y alto riesgo de CG, pancreático, de intestino delgado, mama, cérvix, útero, ovario, testículo y pulmón51. Las variantes constitucionales patogénicas se identifican en casi el 90% de los pacientes que cumplen criterios diagnósticos, y son principalmente de tipo truncante.
Síndrome de la poliposis juvenil (PJ, OMIM#174900) causada por variantes patogénicas en BMPR1A y SMAD4. Estos 2 genes codifican proteínas que participan en la ruta de señalización TGF-B/proteína morfogenética del hueso (BMP), que controla el crecimiento de las células epiteliales del colon, además de modular procesos de diferenciación, proliferación o adhesión celular. BPMR1A es un receptor de membrana celular mientras que SMAD4 es un mediador intracelular de la ruta TGF-B. En el 50-70% de los individuos afectos se identifican variantes patogénicas en BMPR1A o en SMAD4. Los individuos portadores tienen alto riesgo de desarrollar cánceres gastrointestinales51. Los portadores de variantes patogénicas en SMAD4 presentan también riesgo de desarrollar telangiectasia hemorrágica hereditaria. Aunque se han descrito variantes patogénicas en el gen ENG, implicado también en la ruta TGF-B, su papel como responsable de Poliposis Juvenil no está bien definido y está recogido como gen de evidencia limitada en ClinGen (ClinGen: Hereditary Cancer Expert Panel: https://search.clinicalgenome.org/kb/genes/HGNC:3349).
Síndrome de tumores hamartomatosos asociado a PTEN (PTEN- hamartoma tumor síndrome; STHP) (OMIM#158350, 158350, 60530) incluye varias entidades clínicas causadas por variantes patogénicas constitucionales en PTEN: síndrome de Cowden, síndrome de Bannayan-Riley-Ruvalcaba y síndromes Proteus y Proteus-like. Este gen supresor de tumores codifica una fosfatasa que participa en la ruta PI3K/AKT/mTOR. Además de CCR, los individuos portadores tienen alto riesgo de presentar tumores benignos y/o malignos del tracto gastrointestinal, tiroides, mama, endometrio y sistema nervioso central, así como lesiones mucocutáneas, características que ayudan al diagnóstico51.
Poliposis mixtas: Son situaciones poco frecuentes que se caracterizan por presentar múltiples pólipos de diferente histología (adenomas, serrados, hamartomas) que se asocian a un incremento del riesgo de desarrollar CCR. En la mayoría no se identifica la causa genética, pero se han identificado familias con una duplicación upstream en GREM1 que incluye la región 3.’ de SCG5, o en algunos casos con variantes patogénicas en BMPR1A8,67,68.
Síndrome de poliposis serrada (SPS): Se define de forma arbitraria por la OMS en base al número, tamaño y localización de los pólipos en el colon y recto. Los criterios diagnósticos vigentes son: 1) ≥5 pólipos serrados proximales al recto, todos ellos ≥5mm, con al menos 2 de ellos >10mm; 2) >20 pólipos serrados de cualquier tamaño, ≥5 proximales al recto8. Se han reportado variantes constitucionales patogénicas en el gen supresor tumoral RNF43 en el 2% de pacientes con SPS. Aunque en algunos individuos que cumplen criterios clínicos de SPS se han identificado variantes patogénicas en MUTYH, genes MMR, PTEN, GREM1 o NTHL1, los resultados del análisis genético son, por lo general, no informativos8,67.
Escenarios clínicos en los que se recomienda el uso de paneles de genes en las poliposis gastrointestinales- •
Se recomienda la realización de panel de genes en los pacientes con múltiples adenomas colorrectales que cumplan las siguientes características:
- ∘
≥20 adenomas a cualquier edad
- ∘
≥10 adenomas antes de los 40 años, asociado a CCR o con alguna de las manifestaciones clínicas asociadas a la PAF
- ∘
≥10 adenomas y familiar de primer grado con >10 pólipos, CCR o con alguna de las manifestaciones extracolónicas asociadas a la PAF
Calidad de la evidencia baja, nivel de recomendación fuerte a favor.
- •
Se recomienda estudio mediante panel de genes en los pacientes con sospecha clínica o fenotípica de poliposis hamartomatosa. Ante una sospecha concreta, se puede centrar el estudio en los genes relacionados.
Calidad de la evidencia baja, nivel de recomendación fuerte a favor
- •
No se recomienda la realización de panel de genes en los pacientes con adenomas duodenales aislados, no asociados a poliposis colorrectal.
Calidad de la evidencia muy baja, nivel de recomendación fuerte en contra
- •
En los pacientes con síndrome de poliposis serrada, se sugiere realizar estudio mediante paneles de genes en pacientes diagnosticados antes de los 50 años o con al menos un familiar de primer grado con poliposis serrada.
Calidad de la evidencia muy baja, nivel de recomendación débil a favor
Genes a incluir en un panel de poliposis hereditariaResumen de la evidencia- –
Se recomienda el uso de paneles de genes que incluyan todos los genes con suficiente evidencia científica de asociación a síndromes de poliposis hereditaria69. Los genes que se han descrito anteriormente son, en su mayoría, genes con suficiente evidencia científica incluidos en ClinGen (https://search.clinicalgenome.org) y Genomics England Panel App (https://panelapp.genomicsengland.co.uk/panels). Para algunos existen discrepancias como la duplicación upstream de GREM1, con evidencia definitiva para poliposis mixta en GlinGen, pero moderada en PanelApp y MBD4 con evidencia moderada en PanelApp, pero todavía en revisión por panel de expertos en ClinGen, debido a su reciente descubrimiento, pero que apunta evidencias para ser considerado gen de interés. Por su parte, MSH3 y AXIN2 están recogidos actualmente como genes de evidencia moderada para poliposis y CCR tanto en ClinGen como en PanelApp.
- •
Se recomienda un panel de genes de poliposis hereditaria que incluya, al menos, todos los genes con fuerte evidencia de asociación: APC, AXIN2a, BMPR1A, EPCAM (deleción E8-E9), GREM1 (duplicación upstream), MBD4a, MLH1, MSH2, MSH3a, MSH6, MUTYH, NTHL1, PMS2, POLD1b, POLEb, PTEN, RNF43, SMAD4, STK11. Además, se recomienda incluir el análisis de los genes BRCA1 y BRCA2 como cribado oportunista.
- a.
La evidencia científica es moderada, por lo que no se consideran estrictamente necesarios para el diagnóstico. Su inclusión podría ayudar a aportar nuevas evidencias.
- b.
Dominio exonucleasa.
En los pacientes con poliposis hamartomatosa, el análisis genético se debe focalizar en los genes asociados a dichos síndromes.
Calidad de la evidencia muy baja, nivel de recomendación fuerte a favor
3. Uso de paneles de genes en cáncer gástricoIntroducción y revisión de la evidenciaEl cáncer gástrico (CG) es el quinto cáncer más frecuente en el mundo y una de las principales causas de muerte oncológica. En España, la incidencia es de 7,8 casos por 100.000 habitantes, siendo dos veces más frecuente en hombres que en mujeres70. A nivel histológico, se clasifica principalmente en adenocarcinoma tipo intestinal y difuso. El CG intestinal se asocia más a factores ambientales y edad avanzada, mientras que el CG difuso ocurre en personas más jóvenes y se caracteriza por un infiltrado multifocal con células en anillo de sello. La etiología del CG es multifactorial, entre los agentes ambientales principales se encuentran la infección por Helicobacter pylori, la dieta y el tabaco. Si bien la mayoría de los CGs son esporádicos, se observa una agregación familiar en aproximadamente el 10% de casos. El CG hereditario, es decir, en contexto de una variante constitucional asociada, representa un 1-5% de todos los CGs70. Entre las características familiares que sugieren una predisposición hereditaria se incluyen el presentar varios familiares afectos, un patrón de herencia autosómico dominante, afectación a edades tempranas y la asociación a otras neoplasias extragástricas.
En base a la historia familiar y personal, así como el tipo histológico del tumor (difuso o intestinal), se valorará si el paciente cumple criterios clínicos diagnósticos de alguna situación con base hereditaria asociada a un mayor riesgo de CG. En caso afirmativo, se realizará el estudio genético constitucional correspondiente. Un estudio reciente71, demostró una rentabilidad diagnóstica de 43,7% para detección de un síndrome hereditario en pacientes con CG que cumplían criterios clínicos sugestivos de un síndrome hereditario.
Agregación familiar de cáncer gástrico: existen principalmente tres situaciones con agregación de CG. Cada una de estas entidades tiene unos criterios diagnósticos específicos.
- •
Cáncer gástrico difuso hereditario (CGDH)72–75: El CGDH representa el 1-3% de todos los CGs. Es una entidad con herencia autosómica dominante, asociada a variantes patogénicas constitucionales en el gen CDH1 y, menos frecuentemente, en CTNNA1. Para evaluar de forma correcta que una familia cumpla los criterios de CGDH es esencial el informe de anatomía patológica, preferiblemente valorado por un patólogo experto en CG. Los casos confirmados de CG de tipo intestinal no forman parte del CGDH, por ello, en estas familias no estaría indicado realizar análisis genético constitucional. Los criterios diagnósticos de sospecha y que indican la realización de estudio genético, de acuerdo a las recomendaciones del International Gastric Cancer Linkage Consortium (IGCLC) del 202075, Se incluyen en la tabla 5. La rentabilidad diagnóstica del estudio genético es del 30% para CDH1 y del 1-2% para CTNNA1, considerando los casos que cumplen criterios de CGDH.
Tabla 5.Síndromes de agregación familiar de cáncer gástrico: criterios diagnósticos
Agregación Criterios clínicos de sospecha Cáncer gástrico hereditario difuso (CGDH)75 – Criterio familiar• Familia con ≥2 CG (1.° o 2.° grado), cualquier edad, al menos 1 CGD confirmado• Al menos un familiar con CGD a cualquier edad, y uno con cáncer de mama lobulillar ≤70 años• 2 casos de cáncer de mama lobulillar ≤50 años– Criterio personal• CGD<50 años• CGD a cualquier edad en individuo de etnia maorí• CGD e historia personal o familiar de labio leporino• Antecedente personal de CGD y cáncer de mama lobulillar, ambos ≤70 años• Ca mama lobulillar bilateral ≤70 años• Carcinoma in situ gástrico con células en anillo de sello y/o extensión pagetoide de células en anillo de sello en individuos <50 años Adenocarcinoma gástrico asociado a poliposis proximal en estómago (AGPPE) – Poliposis gástrica de cuerpo o fundus sin evidencia de poliposis colorrectal o duodenal y,– 100 pólipos tapizando el estómago proximal en el caso índice o >30 pólipos en un FPG de un paciente con AGPPE y,– Predominio de pólipos fúndicos, algunos con displasia y,– Un familiar con pólipos fúndicos displásicos o CG, y– Patrón autosómico dominante Cáncer gástrico familiar – 3 FPG o FSG con CG, con independencia de la edad, o– 2 FPG o FSG con CG, al menos uno afecto antes de los 50 años
El labio leporino y el paladar hendido son malformaciones congénitas de causa genética en el 30% de los sujetos con implicación de diferentes genes en la etapa del desarrollo orofacial entre ellos CDH1 Las series de casos publicados de CGDH son pequeñas y con una gran variabilidad fenotípica. Específicamente en lo referente al labio leporino, existen dos estudios que describen un 7% de sujetos con labio leporino en portadores de mutación CDH1 en línea constitucional en 2 familias holandesas y un 6% en otras 2 familias francesas. En base a esto, se concluye que, en familias con sospecha de CG difuso, se deben buscar de forma rigurosa y sistemática antecedentes personales y familiares de labio leporino76,77. Por tanto, además del CG difuso y el cáncer de mama lobulillar, malformaciones congénitas en recién nacidos como labio leporino/paladar hendido pueden hacer sospechar un síndrome de CGDH de cara a evaluar un estudio genético.
- –
Adenocarcinoma gástrico asociado a poliposis proximal en estómago (AGPPE)78,79: Es un síndrome recientemente descrito de poliposis gástrica proximal con un riesgo elevado de CG tipo intestinal. La primera familia fue identificada en Australia y, posteriormente, se han identificado otras familias en EE. UU. y Canadá. Los pacientes presentan típicamente poliposis de glándulas fúndicas con áreas de displasia o adenocarcinoma gástrico tipo intestinal restringido al estómago proximal y sin evidencia de poliposis duodenal ni colónica. La edad de presentación del CG es variable, con una media de 50 años (se han identificado casos desde los 23 hasta los 75 años) y con un efecto de anticipación78. Este síndrome tiene una herencia autosómica dominante, con penetrancia incompleta y se ha asociado a variantes patogénicas puntuales en el promotor 1B del gen APC (se considera una variante de la PAF)79. Los criterios clínicos se enumeran en la tabla 5. Se debe distinguir de otros síndromes hereditarios como la PAF clásica o atenuada, la PAM y el síndrome de Peutz-Jeghers. El uso de inhibidores de la bomba de protones también forma parte del diagnóstico diferencial, por lo cual, para hacer el diagnóstico de AGPPE, se debe excluir el uso de estos y repetir la endoscopia tras suspender este tratamiento.
- –
Cáncer gástrico familiar (CGF)54,80–82: Esta entidad hace referencia a familias con agregación de CG de histología intestinal sin causa genética constitucional asociada. En países con alta incidencia de CG, los criterios diagnósticos para CGF son análogos a los criterios de Ámsterdam para el síndrome de Lynch. Sin embargo, en países de baja incidencia de CG, como es España, los criterios clínicos son menos estrictos (tabla 5). La causa genética subyacente se desconoce y no hay consenso sobre la rentabilidad diagnóstica del estudio genético constitucional. De hecho, estudios recientes plantean la posibilidad de una causa poligénica. Se estima que los familiares de primer grado de los pacientes con CG tienen un riesgo de presentar esta neoplasia incrementado 2-3 veces en comparación con la población general. Este riesgo aumenta en base al número de familiares afectos, pudiendo ser de hasta 10 veces más si se tienen 2 o más familiares de primer grado afectos. La edad media de presentación es de 72 años70,80.
Cáncer gástrico y antecedente personal de otros tumores (mama, ovario, colon, endometrio): el CG puede estar presente en diferentes síndromes hereditarios con predisposición a desarrollar tumores en múltiples ubicaciones: síndrome de cáncer de mama y ovario hereditarios (SCMOH), síndrome de Li-Fraumeni, síndrome de Lynch, PAF, PAM, PJ, SPJ o STHP72,83–85. El riesgo a lo largo de la vida de desarrollar CG en estos síndromes es muy variable, pudiendo ser elevado en PJ, SPJ (10-30%)86,87 y probablemente poco frecuente en PAF, LF, PAM, STHP y SCMOH, dependiendo de factores ambientales y geográficos88 (tabla 6). Debemos sospechar alguno de estos síndromes en pacientes con CG, sea de histología intestinal o difusa, que presentan otros tumores asociados. Existe gran variabilidad en la expresión y, debido al solapamiento fenotípico, actualmente la mejor forma de abordar el CG asociado a otros tumores es mediante el uso de paneles de genes, incluyendo genes responsables del síndrome que se sospeche. En una revisión encontraron, en 71 familias estudiadas con criterios de CGHD y CG familiar, 28 variantes patogénicas en 16 genes, siendo las más frecuentes en PALB2 y BRCA2. Solo 5 probandos de estas familias con CG presentaban otro tumor, siendo el más frecuente el cáncer de mama89. Hall et al. analizaron el espectro de neoplasias en portadores de variantes patogénicas en ATM (n = 4.607), encontrando un riesgo moderado-alto de CG (OR: 2,97; IC 95%: 1,66-5,31)90.
Riesgo acumulado de cáncer gástrico en los diferentes síndromes hereditarios asociados
| Síndrome | Gen | Riesgo acumulado |
|---|---|---|
| Síndrome de Peutz-Jeghers | STK11 | 30% |
| Síndrome de Lynch | MLH1/MSH2/MSH6/PMS2 | 11-19% |
| PAF | APC | 0,5-2% |
| SCMOH | BRCA1/BRCA2/PALB2/ATM | 2-5% |
| Síndrome de Li-Fraumeni | TP53 | 2-3% |
| PAM | MUTYH | Bajo, no estimado |
| Poliposis juvenil | SMAD4/BMPR1A/ENG | 21% |
| Síndrome de Cowden | PTEN | Bajo, no estimado |
PAF: poliposis adenomatosa familiar; PAM: poliposis adenomatosa asociada a MUTYH; SCMOH: síndrome de cáncer de mama y ovario hereditarios.
Fuente: adaptado de Setia et al.88.
Cáncer gástrico de aparición precoz: la edad media al diagnóstico del CG es de 60 años, tan solo un 7% ocurren antes de los 50 años y un 2% antes de los 40 años71,91. Sin embargo, aunque la incidencia de CG está disminuyendo a nivel mundial, el CG de aparición precoz aumenta. De hecho, en EE. UU., un estudio reciente demostró que hoy el CG de aparición precoz representa hasta el 30% de todos los CG. La definición de CG de aparición temprana varía según los estudios, pero una de las definiciones más aceptadas incluye a aquellos diagnosticados a los 50 años o antes.
El CG de aparición precoz se ha asociado con algunas alteraciones clínicas y patológicas, como predominio de histología difusa (en torno al 70-75% de los casos, según estudios recientes) y asociación infrecuente con metaplasia intestinal, más frecuentemente estadio avanzado al diagnóstico y, por tanto, alta mortalidad71,92. Con respecto a la edad al diagnóstico del CG para indicar un estudio genético, en el CG de histología difusa está bien establecido en todos los menores de 50 años, independientemente de cualquier otro factor asociado, siendo uno de los criterios del CGDH en contexto de variante patogénica de CDH1 y CTNNA183,92,93.
Con respecto al CG de tipo intestinal, no hay evidencia que respalde un punto de corte para realizar estudio genético y en la mayoría de los síndromes hereditarios asociados, el CG se presenta en mayores de 50 años. Un estudio reciente evaluó la rentabilidad diagnóstica del estudio genético en pacientes jóvenes con CG, evidenciando que solo el 15% de los que presentaban una variante patogénica constitucional tenían antecedentes familiares de CG, pero que la agregación familiar de otros tumores asociados a estos síndromes hereditarios era frecuente91. Así, los criterios más determinantes para identificar un síndrome hereditario asociado eran la edad de diagnóstico antes de los 40 años y la historia personal o familiar de otra neoplasia asociada a dicho síndrome hereditario71,72.
Escenarios clínicos en los que se recomienda el uso de paneles de genes en el cáncer gástrico- •
En individuos con agregación familiar de CG, se recomienda realizar estudio genético constitucional si se cumple criterio de CGDH o AGPPE.
Calidad de la evidencia baja, nivel de recomendación fuerte a favor.
- •
No se recomienda realizar estudio genético constitucional en familias que cumplen criterios de CGF, fuera de estudios de investigación.
Calidad de la evidencia baja, nivel de recomendación fuerte en contra.
- •
Se recomienda estudio constitucional en pacientes con CG y:
- ∘
Poliposis gastrointestinal.
- ∘
Neoplasia del espectro del síndrome de Lynch: colon, endometrio, ovario, páncreas, urotelio, intestino delgado, adenoma sebáceo, tumor del SNC.
Calidad de la evidencia baja, nivel de recomendación fuerte a favor.
- •
En pacientes con CG y antecedente personal o familiar de cáncer de mama y/u ovario, se recomienda analizar los genes asociados a SCMOH solo cuando se cumplen los criterios que definen dicho síndrome.
Calidad de la evidencia baja, nivel de recomendación fuerte a favor.
- •
Se recomienda realizar estudio genético constitucional en pacientes de ≤40 años con CG intestinal o de ≤50 años con CG difuso.
Calidad de la evidencia baja, nivel de recomendación fuerte a favor.
- •
En las familias con CG difuso se recomienda buscar antecedentes de labio leporino/paladar hendido.
Calidad de la evidencia baja, nivel de recomendación fuerte a favor.
- •
Se sugiere realizar estudio genético constitucional si se detecta una asociación familiar y/o personal de CGD y labio leporino/paladar hendido.
Calidad de la evidencia muy baja, nivel de recomendación débil a favor.
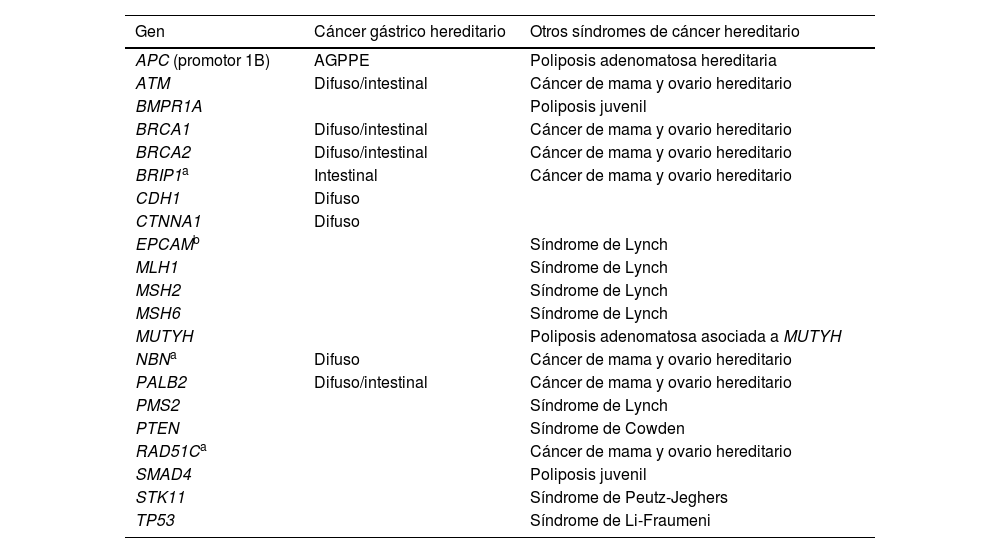
Genes a incluir en un panel de cáncer gástrico hereditarioResumen de la evidenciaSe consideran genes relevantes para la evaluación genética del CG hereditario aquellos relacionados con los síndromes asociados a CG y los síndromes que incluyen el CG dentro de su espectro, tal como se recoge en la tabla 7.
Genes considerados de interés para el diagnóstico genético de cáncer gástrico hereditario
| Gen | Cáncer gástrico hereditario | Otros síndromes de cáncer hereditario |
|---|---|---|
| APC (promotor 1B) | AGPPE | Poliposis adenomatosa hereditaria |
| ATM | Difuso/intestinal | Cáncer de mama y ovario hereditario |
| BMPR1A | Poliposis juvenil | |
| BRCA1 | Difuso/intestinal | Cáncer de mama y ovario hereditario |
| BRCA2 | Difuso/intestinal | Cáncer de mama y ovario hereditario |
| BRIP1a | Intestinal | Cáncer de mama y ovario hereditario |
| CDH1 | Difuso | |
| CTNNA1 | Difuso | |
| EPCAMb | Síndrome de Lynch | |
| MLH1 | Síndrome de Lynch | |
| MSH2 | Síndrome de Lynch | |
| MSH6 | Síndrome de Lynch | |
| MUTYH | Poliposis adenomatosa asociada a MUTYH | |
| NBNa | Difuso | Cáncer de mama y ovario hereditario |
| PALB2 | Difuso/intestinal | Cáncer de mama y ovario hereditario |
| PMS2 | Síndrome de Lynch | |
| PTEN | Síndrome de Cowden | |
| RAD51Ca | Cáncer de mama y ovario hereditario | |
| SMAD4 | Poliposis juvenil | |
| STK11 | Síndrome de Peutz-Jeghers | |
| TP53 | Síndrome de Li-Fraumeni |
La evidencia científica de la implicación de los genes BRIP1, NBN y RAD51C en cáncer gástrico es moderada, por lo que a día de hoy no se considera imprescindible la inclusión de estos genes en el diagnóstico. Su inclusión en el panel de genes de cáncer gástrico podría ayudar a aportar nuevas evidencias para consolidar su relación causa-efecto.
Se recomienda incluir en un panel de genes para el estudio de cáncer gástrico hereditario los siguientes genes: CDH1, CTNNA1, APC (incluyendo variantes puntuales en promotor 1B), ATM, BMPR1A, BRCA1, BRCA2, BRIP1a, EPCAM (deleción E8-E9), MLH1, MSH2, MSH6, MUTYH, NBNa, PALB2, PMS2, PTEN, RAD51Ca, SMAD4, STK11, TP53.
Calidad de la evidencia baja, nivel de recomendación fuerte a favor.
a La evidencia científica de la implicación de los genes BRIP1, NBN y RAD51C en CG es moderada, por lo que a día de hoy estos genes no se consideran estrictamente necesarios para el diagnóstico. No obstante, su inclusión en el panel de genes de CG podría ayudar a aportar nuevas evidencias para consolidar su relación causa-efecto.
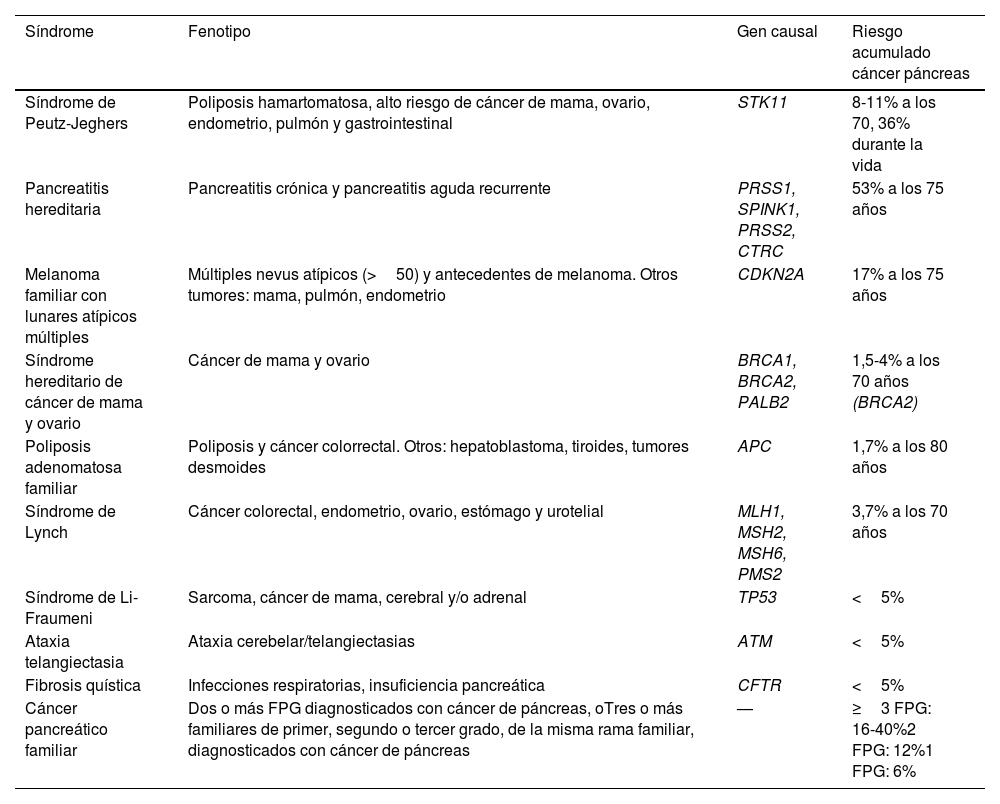
4. Uso de paneles de genes en cáncer de páncreasIntroducción y revisión de la evidenciaEl desarrollo del cáncer de páncreas (CP) se ha relacionado generalmente con factores ambientales, como tabaquismo y consumo de alcohol o enfermedades como la diabetes o la pancreatitis crónica. Pero, aunque la mayoría de los CP son esporádicos, aproximadamente el 10% presentan una agregación familiar. No se ha identificado ningún gen principal causante de cáncer de páncreas, pero las variantes patogénicas en varios genes están relacionadas con un mayor riesgo de este tumor dentro de síndromes de predisposición a diversos tumores (tabla 8). Estos síndromes de cáncer hereditarios representan el 3% de todos los casos de CP. Por otro lado, en el 7% de los casos de cáncer de páncreas existe una fuerte historia familiar sin una variante constitucional causante, situación conocida como cáncer pancreático familiar (CPF)94. El CP tiene 2 factores que determinan la utilización o no de paneles de genes, por un lado, la baja incidencia de la enfermedad (8 casos por 100.000 habitantes/año) y, por otro, la poca eficacia de los tratamientos que determina una alta mortalidad (menos del 10% sobrevive a los 5 años del diagnóstico), Por ello, es clave la identificación de individuos con alto riesgo de desarrollarlo, bien por la presencia de factores de riesgo y enfermedades, o por tener predisposición genética, a través de paneles de genes. Todo ello tiene como objetivo el cribado de estas personas y la detección de tumores en fases precoces.
Síndromes hereditarios/familiares asociados a incremento de riesgo de cáncer de páncreas
| Síndrome | Fenotipo | Gen causal | Riesgo acumulado cáncer páncreas |
|---|---|---|---|
| Síndrome de Peutz-Jeghers | Poliposis hamartomatosa, alto riesgo de cáncer de mama, ovario, endometrio, pulmón y gastrointestinal | STK11 | 8-11% a los 70, 36% durante la vida |
| Pancreatitis hereditaria | Pancreatitis crónica y pancreatitis aguda recurrente | PRSS1, SPINK1, PRSS2, CTRC | 53% a los 75 años |
| Melanoma familiar con lunares atípicos múltiples | Múltiples nevus atípicos (>50) y antecedentes de melanoma. Otros tumores: mama, pulmón, endometrio | CDKN2A | 17% a los 75 años |
| Síndrome hereditario de cáncer de mama y ovario | Cáncer de mama y ovario | BRCA1, BRCA2, PALB2 | 1,5-4% a los 70 años (BRCA2) |
| Poliposis adenomatosa familiar | Poliposis y cáncer colorrectal. Otros: hepatoblastoma, tiroides, tumores desmoides | APC | 1,7% a los 80 años |
| Síndrome de Lynch | Cáncer colorectal, endometrio, ovario, estómago y urotelial | MLH1, MSH2, MSH6, PMS2 | 3,7% a los 70 años |
| Síndrome de Li-Fraumeni | Sarcoma, cáncer de mama, cerebral y/o adrenal | TP53 | <5% |
| Ataxia telangiectasia | Ataxia cerebelar/telangiectasias | ATM | <5% |
| Fibrosis quística | Infecciones respiratorias, insuficiencia pancreática | CFTR | <5% |
| Cáncer pancreático familiar | Dos o más FPG diagnosticados con cáncer de páncreas, oTres o más familiares de primer, segundo o tercer grado, de la misma rama familiar, diagnosticados con cáncer de páncreas | — | ≥3 FPG: 16-40%2 FPG: 12%1 FPG: 6% |
Fuente: adaptado de Llach et al.94.
Análisis genético en pacientes con CP sin historia personal y/o familiar de otros cánceres: tampoco existe una evidencia sólida sobre cuándo solicitar un panel de genes en pacientes con CP sin otros antecedentes. La edad media de diagnóstico del CP se sitúa en la séptima década de la vida en la mayoría de los estudios. En tres estudios se valoró la presencia de variantes genéticas patogénicas en menores de 50 años con CP y no se encontraron diferencias con el resto de grupos de edad95–97. Algunos consensos habían establecido los 45 años para indicar el estudio con un panel de genes. Sin embargo, la tendencia es a elevar el punto de corte hasta los 60 años de edad98. Como se ha comentado anteriormente, se ha sugerido generalizar el estudio genético a todos los pacientes que tienen metástasis, por la posibilidad de ofrecer tratamientos más efectivos en aquellos que tienen variantes patogénicas constitucionales, como por ejemplo en BRCA1 y BRCA2, que responden mejor a cisplatino e inhibidores de PARP. En base a los estudios realizados hasta este momento, parece razonable no extender la utilización de paneles de genes a todos los pacientes con CP, y solo realizarlos en aquellos que debutan a una edad inferior a los 60 años, siendo este punto de corte arbitrario.
Análisis genético en pacientes con CP y antecedente personal/familiar de otros tumores (mama, próstata, melanoma, colon): las publicaciones disponibles demuestran un incremento de variantes patogénicas detectadas en este grupo de pacientes y esta evidencia ha quedado reflejada en la mayoría de los consensos publicados99–101. Recomendamos que se realice el estudio genético en todos los pacientes con CP y antecedente personal de otros tumores (mama, próstata, melanoma, colon). En todos los casos en los que un paciente tenga un CP y tenga un familiar de primer grado (FPG) con alguno de estos tumores se debe analizar un panel de genes. El tener un FPG afecto de cáncer de mama, próstata, melanoma o colon incrementa 2,36 veces la probabilidad de detectar variantes patogénicas102. En este caso el porcentaje de casos detectados es superior al 10 frente al 2,5% si no hay ningún antecedente personal o familiar de cáncer (tabla suplementaria 1)100,102,103,129. En otros estudios, a los tumores mencionados anteriormente se asocian también vejiga, ovario y útero99.
Análisis genético en pacientes con CP y agregación familiar de CP: el CPF representa el síndrome más frecuente de adenocarcinoma pancreático exocrino, en el cual se observa una agregación familiar de esta neoplasia sin una causa genética identificada (80-90% de los casos). El incremento del riesgo de CP está condicionado por el número de familiares afectos104. De tal modo que la afectación de un FPG hace que el riesgo acumulado pase de un 1-2% de la población general a un 4-6%, si existen 2 FPG el riesgo sube al 7%, y si son 3 FPG llega hasta el 17-32%99,101,104,129. Cuando se cumplen criterios de CPF (2 o más familiares de primer grado diagnosticados con CP o 3 o más familiares de primer, segundo o tercer grado, de la misma rama familiar, diagnosticados con CP) la probabilidad de detectar variantes patogénicas constitucionales incrementa de forma notable, llegando al 14%99,105,106. Las mutaciones más frecuentes se encuentran en BRCA1, BRCA2, ATM, CDKN2A, PALB2, PTEN, APC y FANC.
Análisis genético en pacientes con lesiones preneoplásicas: en el momento actual hay pocos estudios que analizan este aspecto y los que hay observan una prevalencia de variantes patogénicas por debajo del 10%. Por ejemplo, un reciente estudio en China107 identifica variantes patogénicas en el 2,8% de los tumores papilares mucinosos intraductales (TPMI) (2 de 70 pacientes: genes RECQL4 y ATM). El paciente con la variante en ATM desarrolló un carcinoma invasivo. En otro estudio se detectaron variantes patogénicas en 9 de 315 (2,9%) de los pacientes con TPMI108. En otro estudio, se observaron variantes patogénicas en 2 de 32 pacientes con tumores serosos (6,3%; BRCA2 y Cystic Fibrosis Transmembrane Receptor [CFTR]) y en en 2 de 56 pacientes con tumores sólidos pseudopapilares (3,6%; PALB2 y ATM). También se encontró una variante patogénica en BRCA1 en un paciente de 119 casos con tumores neuroendocrinos de páncreas (0,8%)109.
Análisis genético en pacientes con pancreatitis no alcohólica: en una cohorte de 98 pacientes con pancreatitis crónica idiopática en China encontraron variantes patogénicas en el 20,4% (20 pacientes), la mayoría en el gen SPINK1 (19 pacientes), siendo la mutación c.194+2T>C la más frecuente, y 1 en PRSS1107. Un paciente presentaba mutaciones en SPINK1 y en BRIP1. La mutación c.194+2T>C en el gen SPINK1 tiene una prevalencia en pacientes con CP en China del 2,1%, frente al 1,1% en la población general. En cambio, en pacientes con pancreatitis aguda idiopática hay pocos estudios y la prevalencia de variantes patogénicas es baja, por lo que no recomendamos al análisis de paneles de genes en este grupo de pacientes. La causa más frecuente en este grupo de pacientes es por barro biliar. En un grupo de pacientes con pancreatitis aguda idiopática se detectaron variantes patogénicas en 15 de 355 pacientes (4,2%). En estos casos, las variantes detectadas afectaban a SPINK1 (11 pacientes, todos con la mutación c.194+2T>C), CFTR (2 pacientes), MLH1 (un paciente) y ATM (un paciente)107. Los genes que con más frecuencia se han asociado con la pancreatitis crónica no alcohólica son SPINK1, PRSS1 y CFTR. No se sabe si estos genes pueden inducir el desarrollo de cáncer de páncreas por sí mismos o es a través del desarrollo de una pancreatitis crónica. La proteína SPINK1 actúa inhibiendo la tripsina pancreática y previene la activación prematura del tripsinógeno. La reducción de su actividad por mutaciones en el gen SPINK1 favorece la activación enzimática y la inflamación. Aunque las mutaciones constitucionales en el gen SPINK1 son muy frecuentes en pacientes con pancreatitis crónica idiopática, su papel como factor de riesgo en el cáncer de páncreas es muy controvertido. En algunos estudios se ha visto que puede incrementar hasta 12 veces el riesgo de desarrollar CP, sin embargo, este factor de riesgo solo lo asocian con la mutación p. N34S(c.101A>G)110.
La presencia de mutaciones bialélicas en el gen CFTR es frecuente en pacientes con pancreatitis crónica. Sin embargo, no existe evidencia sobre su asociación con el CP111. El gen CFTR está íntimamente relacionado con la fibrosis quística. La fibrosis quística es una enfermedad hereditaria, autosómica recesiva, con una frecuencia de un caso cada 2.000-3.000 nacimientos. La prevalencia de portadores de mutaciones en CFTR es del 3 al 5%. Un 85% de los pacientes con fibrosis quística clásica desarrolla insuficiencia pancreática exocrina; sin embargo, no se ha asociado a un incremento del CP.
El gen PRSS1 o gen de la proteasa de serina 1 o del tripsinógeno catiónico, codifica la tripsina catiónica que interviene en la activación del tripsinógeno a tripsina112. Alrededor de un 8% de las pancreatitis crónicas son debidas a mutaciones genéticas en PRSS1. Se debe solicitar el análisis de este gen si hay 2 o más pacientes en dos o más generaciones de una familia con pancreatitis recurrentes de causa idiopática por debajo de los 25 años. Se han descrito más de 20 mutaciones en este gen. El riesgo de CP en los pacientes con pancreatitis hereditaria es muy elevado, independientemente de la presencia o no de mutaciones del gen PRSS1, aconsejando cribado a partir de los 40 años o pasados 20 años de la primera pancreatitis. El riesgo del cáncer de páncreas viene dado por la presencia de pancreatitis crónica.
Escenarios clínicos en los que se recomienda el uso de paneles de genes en el CP- •
Se sugiere el análisis de un panel de genes en pacientes con cáncer de páncreas diagnosticado antes de los 60 años independientemente de los antecedentes familiares o personales.
Calidad de la evidencia baja, nivel de recomendación débil a favor.
- •
Se sugiere el análisis de un panel de genes en los pacientes con cáncer de páncreas y antecedente personal y/o familiar de otros tumores (mama, próstata, melanoma o colon).
Calidad de la evidencia baja, nivel de recomendación débil a favor.
- •
Se recomienda la realización de panel de genes en los pacientes con cáncer de páncreas que cumplen criterios de cáncer de páncreas familiar.
Calidad de la evidencia baja, nivel de recomendación fuerte a favor.
- •
No se recomienda realizar análisis constitucional mediante panel de genes en pacientes con lesiones preneoplásicas.
Calidad de la evidencia baja, nivel de recomendación fuerte en contra.
Genes a incluir en un panel de cáncer de páncreas hereditarioLos genes en los que se detectan variantes patogénicas con mayor frecuencia en CP son BRCA1, BRCA2, MLH1, MSH2, MSH6, ATM, PALB2 y CDKN2A y PRSS1 (en el caso de pancreatitis crónica idiopática). Este grupo de genes ha sido asumido por el grupo de expertos del proyecto IMPaCT Genómica3. Otros genes que se incluyen en diferentes estudios son TP53, CFTR (en casos de sospecha de fibrosis quística), PMS2, y EPCAM. El síndrome de Li-Fraumeni es una enfermedad autosómica dominante producida por variantes patogénicas en el gen TP53, proteína vital para regular el ciclo celular y el reconocimiento de daños en el ADN. Ciertas mutaciones constitucionales en este gen se han asociado a un incremento del riesgo de CP110. En caso de presentar poliposis se podría escoger otro tipo de genes o paneles que incluya APC, MUTYH, STK11, SMAD4, POLE, POLD1 y PTEN.
Recomendación:- –
Se recomienda incluir en un panel de genes aquellos asociados a predisposición de CP: BRCA1, BRCA2, MLH1, MSH2, MSH6, ATM, PALB2, CDKN2A, PRSS1a, TP53, CFTRb, PMS2 y EPCAM. Si el CP se asocia en el paciente o la familia a poliposis intestinal se incluirán APC, MUTYH, STK11, SMAD4, POLE, POLD1 y PTEN.
- a.
Si pancreatitis crónica idiopática.
- b.
Si sospecha de fibrosis quística.
Calidad de la evidencia baja, nivel de recomendación fuerte a favor.
5. Estudio de mosaicismosIntroducción y resumen de la evidenciaLas variantes genéticas que se encuentran en las células germinales (óvulo o espermatozoide) de los progenitores y que, por tanto, pueden ser transmitidas a la descendencia se denominan variantes hereditarias. En caso de heredarse, estas variantes estarían presentes en todas las células del organismo (variante genética constitucional). Por el contrario, las variantes genéticas somáticas surgen en alguna etapa de la vida en tejidos no germinales y no se heredan. Ambas definiciones anteriormente expuestas presentan matices, pues existen individuos que presentan una variante genética constitucional pero que no está presente en células somáticas (p. ej., ADN de leucocitos) de ninguno de los progenitores, habiendo descartado falsa paternidad o maternidad. Este tipo de variación genética, que aparece por primera vez en la familia, se denomina variante de novo, y puede surgir de dos formas: 1) la alteración genética está presente en alguna de las células germinales de uno de los progenitores, pero no en el resto de sus células (mosaicismo germinal); en este caso un progenitor que no presenta una patología puede transmitir la condición genética a su descendencia; o 2) el cambio genético tiene lugar en una fase temprana del desarrollo embrionario (óvulo ya fertilizado), pero no está presente en ninguna de las células germinales de los progenitores (mosaicismo somático). En este caso el individuo presenta un conjunto de células con una composición genética distinta al resto. Esta mutación temprana ha dado lugar a una población de células hijas que portan la misma alteración genética, mientras que el resto de las células del organismo (procedentes de otras células sin la alteración) no la presentan. Según el tipo de variante genética y del número de células que presenten la alteración, un mosaicismo somático puede, o no, causar una enfermedad.
Estudio de mosaicismos en caso de poliposis colorrectal: hasta el 20% de los casos de PAF se deben a variantes de novo (sin historia familiar previa) causados principalmente por mosaicismos somáticos en el gen APC, que surgen en distintas etapas de la embriogénesis del individuo y, por tanto, no se han heredado de los progenitores113,114. Esta situación debemos sospecharla ante un caso de poliposis clásica único en la familia y en el que se identifica una variante constitucional patogénica de APC no presente en el ADN de leucocitos de ninguno de los progenitores [habiendo descartado falsa paternidad o maternidad (donación de óvulo, adopción no informada). Si, por el contrario, en este caso aislado de poliposis en la familia no se identifica ninguna variante constitucional patogénica en APC u otros genes asociados a poliposis gastrointestinal, debe considerarse el análisis de ADN, idealmente, de la mucosa sana y de 2 o 3 adenomas, para descartar mosaicismo somático115,116. Si una misma variante patogénica de APC se identifica en varios adenomas se confirmaría el mosaicismo somático113,117. Además de APC, también se ha descrito mosaicismo en otros genes de poliposis como PTEN o STK11, pero, a diferencia de en el caso de APC, son eventos raros.
Recomendaciones:- -
En casos aislados de poliposis adenomatosa familiar clásica con estudio genético en sangre no informativo, se sugiere realizar análisis del gen APC en el ADN de 2-3 adenomas y mucosa sana de colon, para descartar mosaicismo somático.
Calidad de la evidencia baja, nivel de recomendación débil a favor.
- -
En caso de identificar una variante genética causal en mosaico, se recomienda su análisis en la descendencia para confirmar su transmisión.
Calidad de la evidencia baja, nivel de recomendación fuerte a favor.
6. Estrategia de asesoramiento genético ante ausencia de sujeto índice.Introducción y resumen de la evidenciaEl estudio genético para la identificación de una alteración que pueda ser la causa de un cáncer digestivo hereditario debe realizarse, siempre que sea posible, en un individuo afecto vivo. Si se dispone de varios familiares afectos vivos, el candidato ideal es aquel que haya sido diagnosticado a edad más joven o afecto por un tipo de neoplasia menos frecuente dentro del espectro de un síndrome de cáncer hereditario. Normalmente, para la realización de estos estudios se utiliza el ADN/ARN extraído de muestras biológicas obtenidas por métodos no invasivos como sangre periférica y, a veces, saliva72,114. Sin embargo, es frecuente que no se disponga de esas muestras de ningún familiar afecto y que la persona que consulta buscando asesoramiento genético sea el familiar en riesgo de unpaciente con cáncer fallecido. En este escenario, las alternativas actualmente disponibles son: el estudio genético en muestras del familiar fallecido (tumor o tejido sano no tumoral), o bien ofrecer el análisis genético constitucional a familiares sanos118.
La posibilidad creciente de estudio genético mediante NGS en muestra tisular nos permite el análisis genético en el probando óptimo. Aunque la muestra idónea es la criopreservada, lo habitual es disponer de tejido incrustado en parafina, lo que limita el análisis. En cualquier caso, es preferible el estudio del ADN de la mucosa o tejido sano no tumoral, ya que cuando el estudio se realiza a partir de ADN de tumor se identifican tanto la alteración genética constitucional como las somáticas en el mismo gen diana. Es importante incidir durante el asesoramiento genético pre-test de estos casos, que tras un resultado no informativo (en el que no se identifica la variante genética causal) de un test genético realizado en muestras de mucosa o tejido sano persiste una probabilidad relativamente alta de la presencia de una alteración genética subyacente que conlleve una predisposición hereditaria o incremento de riesgo a desarrollar cáncer en la familia. Por otro lado, si se identifica una alteración genética causal a partir de ADN de mucosa sana de un familiar afecto fallecido, se debe validar confirmando la presencia de dicha alteración en sangre de alguno de los familiares del individuo estudiado. Si se identifica una alteración genética es recomendable estudiar la presencia de esa alteración identificada en familiares de primer y segundo grado para su confirmación y la consecuente predicción de riesgo genético en estos.
Si el tejido de mucosa sana no está disponible o no se consigue extraer de esa muestra ADN de suficiente calidad para realizar estudios genéticos, se puede plantear el estudio genético en familiares sanos. La probabilidad de identificar una alteración genética que cause un incremento de riesgo a cáncer en un individuo sano perteneciente a una familia con sospecha de predisposición hereditaria a cáncer y sin diagnóstico genético es inversamente proporcional al grado de consanguinidad con un individuo afecto. Solo en el caso excepcional de que el afecto no disponible tuviese un gemelo univitelino sano, este sería el mejor candidato a estudio, ya que la probabilidad de identificar la alteración genética causal sería la misma que en el hermano afecto. Cuando el individuo sano que se pretende estudiar es familiar de primer grado de un individuo afecto, la probabilidad de identificar la alteración genética causal es un 50% menor que la probabilidad de encontrarla en el afecto. Si es de segundo grado, un 25% y de tercer grado, un 12,5%. Por ejemplo, la probabilidad de encontrar una alteración genética casual en una familia que cumple criterios de CGDH varía entre un 34-54%118. El estudio genético completo de un individuo sano familiar de primer grado de un afecto no disponible tendría un rendimiento diagnóstico teórico de la mitad, es decir, entre el 17-27%, y este rendimiento sería de un 8,5-13,5% para familiares de segundo grado.
En algunos escenarios de predisposición hereditaria al desarrollo de cáncer, como es el SCMOH, se ha establecido la posibilidad de estudio genético de familiares sanos cuando la probabilidad estimada de que en la familia exista un gen alterado sea mayor al 10%.118 Sin embargo, cabe recordar que apenas existen herramientas validadas para estimar la probabilidad de detectar una alteración genética causal de síndromes de predisposición hereditaria a cáncer más allá del SCMOH, el síndrome de Lynch119 o el de Cowden. En el caso del CP, la complejidad en la evaluación del riesgo de cáncer ha llevado al desarrollo de un modelo de predicción de riesgo (PancPRO) para proporcionar estimaciones más detalladas para individuos de familias con CPF en base a las edades en el momento del diagnóstico, el tamaño de la familia y la relación entre los miembros de esta120, y se podría utilizar para refinar la indicación. No obstante, esta herramienta fue publicada en 2007 y, desde entonces, muy pocos estudios la han avalado. En ausencia de estas herramientas, la información sobre la proporción de probandos con variantes patogénicas detectables en familias que cumplen los criterios clínicos de sospecha establecidos, obtenidas de GeneReviews o de PubMed, pueden ser de utilidad para el cálculo de la probabilidad de detectar una variante causal en familiares de primer grado114. En el caso de tener que recurrir a un familiar sano es importante tener en cuenta la edad del familiar. Siempre que sea posible, es preferible estudiar a un familiar sano que tenga una edad igual o menor a la edad al diagnóstico del caso índice natural de la familia, asumiendo que la probabilidad de identificar una alteración genética causal será mayor en un familiar sano de primer grado joven que en otro de edad más avanzada. En cualquier caso, el estudio genético para determinar el riesgo de familiares sanos debe ser interpretado con mucha precaución. Si al analizar al familiar sano se identifica la variante patogénica en el gen responsable del síndrome que se sospecha, se puede ofrecer estudio genético dirigido de esa variante al resto de los familiares sanos en riesgo. Sin embargo, cuando el resultado no es informativo, aún existe una probabilidad elevada de la presencia de una alteración genética subyacente responsable de un incremento de riesgo a cáncer en el individuo o sus familiares.
Recomendaciones- -
Ante la sospecha de cáncer digestivo hereditario sin un individuo afecto vivo y disponible, se recomienda el análisis genético con panel de genes en el tejido sano no tumoral del individuo afecto.
Calidad de la evidencia baja, nivel de recomendación fuerte a favor.
- -
Si se identifica una variante patogénica en tejido sano se recomienda el estudio de familiares de primer y segundo grado para confirmar la presencia de la variante.
Calidad de la evidencia baja, nivel de recomendación fuerte a favor.
- -
En familias que cumplan criterios clínicos de alto riesgo de cáncer hereditario sin disponibilidad de muestras de un sujeto índice, se sugiere la valoración del análisis genético en familiares de primer grado sanos de manera individualizada.
Calidad de la evidencia baja, nivel de recomendación débil a favor.
7. Estrategia de análisis constitucional tras identificación de variante patogénica en secuenciación tumoralEl estudio de variantes patogénicas somáticas mediante secuenciación masiva (NGS) a partir de la muestra tumoral se está convirtiendo en un procedimiento diagnóstico cada vez más común en la era de la medicina de precisión. El objetivo fundamental de estos paneles somáticos es poder identificar alteraciones genéticas específicas que puedan ser diana para determinados fármacos. Por ejemplo, actualmente, en el estudio molecular mediante NGS en pacientes con CG metastásico, ya se recomienda incluir biomarcadores moleculares predictivos de respuesta a tratamiento como (gen [tipo alteración. Nivel ESCAT] ERBB2 (amplificaciones. IA); NTRK (fusiones. IC); EGFR (IIB); MET (amplificaciones. IIB), además de la IMS121,122.
En la práctica clínica es frecuente la utilización de paneles de genes pan-cáncer, que incluyen, además de los biomarcadores recomendados, un número variable de otros genes, ofreciendo una mejora en la eficiencia y en el tiempo de respuesta de los laboratorios que realizan dichos estudios. En una buena parte de estos paneles NGS pan-cáncer se incluyen genes responsables de diferentes síndromes de predisposición hereditaria a cáncer. Por tanto, en el estudio molecular de tumores para la identificación de biomarcadores predictivos de respuesta a tratamientos o marcadores pronósticos mediante NGS, se pueden identificar variantes que, si reúnen determinadas características, pueden sugerir la existencia de una variante patogénica constitucional responsable de un síndrome de cáncer hereditario. Para confirmarlo se requerirá ratificar en ADN de sangre la presencia de la alteración detectada en el tumor. De esta forma, el estudio molecular de tumores puede ser utilizado como un cribado oportunista para la identificación de casos con cáncer hereditario.
La indicación del estudio genético constitucional a partir de variantes detectadas en el estudio molecular del tumor viene determinada por criterios de tipo analítico y clínico:
Entre los criterios analíticos que deben cumplir estas variantes para ser consideradas sospechosas de un síndrome de cáncer hereditario son:
- a)
La variante debe localizarse en un gen reconocido asociado a cáncer hereditario123.
- b)
La variante debe ser clasificada como patogénica o probablemente patogénica según los criterios del ACMG1.
- c)
La frecuencia alélica de la variante (VAF) ha de ser mayor o igual del 30% para variantes de tipo Single Nucleotide Variant (SNV) o sustituciones, o mayor o igual del 20% para variantes de tipo indel124.
Desde el punto de vista clínico, hay que considerar la historia personal y familiar de cáncer (tipos de tumores y edades al diagnóstico) para establecer la indicación del estudio en línea constitucional. Previamente, el paciente debe haber firmado el preceptivo consentimiento informado donde se establezca la gestión de la información obtenida de los estudios genéticos, incluyendo su deseo de conocer o no los resultados que pudieran tener una trascendencia a nivel hereditario. El consentimiento informado determina la forma de actuar en cada caso. Existen estudios en los que se realiza una aproximación de análisis genético pareado de tejido tumoral-normal. Esta estrategia es la que ofrece un mayor rendimiento diagnóstico de síndromes de predisposición hereditaria a cáncer. La principal limitación de este abordaje es su elevado coste y la discutible eficiencia del proceso. El uso de estrategias de cribado oportunista debe demostrar su coste-eficacia125,126.
De acuerdo con las recomendaciones de la European Society of Medical Oncology127, las indicaciones de estudio constitucional cuando se identifica una variante patogénica en la secuenciación de un tumor sólido, siguiendo una estrategia «conservadora intermedia» son:
- •
Variantes con frecuencia alélica (VAF) superior al 30% si se trata de una SNV (sustituciones) o mayor o igual a 20% en variantes tipo indels en los genes BRCA1, BRCA2, PALB2, MLH1, MSH2, MSH6 y RET, en cualquier tumor.
- •
VAF≥30% (sustituciones) o ≥20% (indels) en los genesTP53, RB1, APC, CDKN2A y SMARCA4 identificadas en pacientes diagnosticados antes de los 30 años de tumores asociados al espectro del síndrome hereditario.
- •
VAF≥30% (sustituciones) o ≥20% (indels) en los genes BRIP1, MUTYH bialélico, PMS2, RAD51C, RAD51D, SDHB, TSC2, VHL, ATM, BAP1, BARD1, CHEK2, DICER1, FH, FLCN, NF1, POLD1, POLE y SDHA identificadas en pacientes con tumores asociados al espectro del síndrome hereditario.
Se recomienda realizar el estudio genético constitucional a partir de la detección de una variante patogénica en un panel de secuenciación tumoral, siempre que el paciente lo haya expresado así en el consentimiento informado, ante la identificación de una variante patogénica con VAF≥30% (sustituciones) o ≥20% (indels) en los genes:
- -
BRCA1, BRCA2, PALB2, MLH1, MSH2, MSH6 y RET en cualquier tumor.
- -
TP53, RB1, APC, CDKN2A y SMARCA4 en pacientes diagnosticados antes de los 30 años de tumores asociados al espectro del síndrome hereditario.
- -
BRIP1, MUTYH bialélico, PMS2, RAD51C, RAD51D, SDHB, TSC2, VHL, ATM, BAP1, BARD1, CHEK2, DICER1, FH, FLCN, NF1, POLD1, POLE y SDHA en tumores asociados al espectro del síndrome hereditario.
Calidad de la evidencia baja, nivel de recomendación fuerte a favor.
FinanciaciónEste documento de posicionamiento no ha contado con financiación externa. Joaquin Cubiella ha recibido una financiación del Instituto de Salud Carlos III a través del proyecto PI21/01771 (cofinanciado por el Fondo Europeo de Desarrollo Regional «Una manera de hacer Europa /Invirtiendo en tu futuro».
A la Comisión de Cáncer Hereditario de la AEGH por sus comentarios al documento final.

