







INTRODUCCIÓN
El trasplante hepático es el principal tratamiento de muchas hepatopatías que inciden en niños. Los progresos en diferentes campos, incluida la técnica quirúrgica, una inmunosupresión más eficaz y un mejor control de las infecciones, permiten que en la actualidad el 90% de los niños trasplantados sobrevivan y tengan una buena calidad de vida.
El trasplante hepático se realiza tras reconocer la progresión de una enfermedad y la carencia de otros medios eficaces de tratamiento. El diagnóstico correcto de la causa de la hepatopatía, el conocimiento de su riesgo estadístico de ocasionar mortalidad a diferentes plazos de tiempo y de los datos clínicos asociados al pronóstico permite establecer con buen criterio el momento adecuado para indicar la realización del trasplante.
En el seguimiento después del trasplante pueden desarrollarse complicaciones generales y del injerto, que siguen generalmente un patrón de aparición relacionado con el tiempo transcurrido desde el trasplante. Las complicaciones en los primeros meses y las ocurridas en el seguimiento a más largo plazo son profundamente distintas y requieren acercamientos preventivos y tratamientos diferentes.
INDICACIONES DE TRASPLANTE HEPÁTICO EN NIÑOS
La indicación de trasplante hepático se realiza ante una enfermedad hepática grave no susceptible de otro tratamiento eficaz, cuando el trasplante le puede proporcionar mejor supervivencia y calidad de vida que con el hígado original.
Hepatopatía «grave» es un concepto variable para cada tipo de enfermedad. Es imprescindible asegurar el diagnóstico etiológico correcto. Cada enfermedad tiene un acercamiento de tratamiento médico, en algunas de ellas total o parcialmente eficaz. Cada patología tiene patrones evolutivos sobre los que se establece el pronóstico vital que es el empleado para estimar la necesidad de trasplante. Algunas hepatopatías son difíciles de evaluar en este sentido, unas veces por inexperiencia debida a su baja incidencia, o por ser el hígado asiento de un defecto que repercute solamente en otros órganos y de una manera poco previsible, o por no ofrecer el trasplante una garantía de que evitará el compromiso severo de otros órganos en el seguimiento.
Atresia biliar extrahepática
La atresia biliar extrahepática (ABE) es la enfermedad hepática severa infantil más frecuente. Ocurre en 1 por cada 8.000 a 10.000 recién nacidos. El 40% de los trasplantes hepáticos realizados en niños en todos los países se deben a esta enfermedad.
La ABE es el resultado de un proceso de colangitis esclerosante de causa desconocida que se inicia en el período perinatal y progresa rápidamente, conduciendo a la obliteración fibrosa de la vía biliar extrahepática y a lesiones hepáticas de fibrosis. El niño manifiesta ictericia y acolia en las primeras semanas de vida. La mayoría de los pacientes se evalúa por primera vez entre el primer y segundo mes ya que su buen estado general inicial retrasa la consulta o la alerta del pediatra. Los datos histológicos a esa edad son ya de fibrosis portal moderada a severa junto a proliferación ductal. La enfermedad no tratada evoluciona a cirrosis desde una edad muy precoz. El niño desarrolla insuficiencia hepática y complicaciones derivadas de hipertensión portal (ascitis, hemorragia digestiva) que causan el fallecimiento antes del año de edad en el 90% de los casos. Menos del 3% de los niños sobrevive 3 años.
La identificación correcta de la ABE en el lactante pequeño, preferiblemente antes de los 60 días de edad, para la aplicación de un tratamiento quirúrgico cambia el pronóstico de manera significativa, a expensas de un grupo de niños (el 50%) que se benefician, mientras que en los demás no se conseguirá modificar la evolución1,2.
La cirugía consiste en una portoenteroanastomosis con alguna de las variantes técnicas a partir de la descrita originalmente por Kasai y va dirigida a permitir el paso de la bilis desde los conductos intrahepáticos que queden permeables desde la salida del hígado (porta hepatis) hasta el intestino. El momento de la cirugía es importante; el niño debe ser menor de 3 meses de edad. Aunque en la bibliografía médica se ha enfatizado la importancia de la precocidad de la cirugía, los estudios de diferentes centros, que consideran niños operados antes de los 75 días de vida, no muestran diferencias significativas en los resultados entre niños operados antes del mes, en el segundo o en el inicio del tercer mes1. La experiencia demuestra, sin embargo, que la operación de estos niños a partir del día 90 rara vez consigue resultados, pues el hígado es cirrótico y la atresia ha progresado al comienzo de la vía intrahepática, lo que impide el restablecimiento de flujo biliar pues no hay conductillos permeables en la porta hepatis.
El pronóstico del paciente no puede establecerse basado en los datos que tiene en el momento de la operación, incluidos su edad, parámetros de laboratorio, estadio de fibrosis o diámetro de los conductos en la porta hepatis seccionada. Ninguno de esos datos, en niños menores de 75 días, se relaciona fiablemente con la evolución. Lo que importa es si en el curso posterior a la cirugía, valorado en un plazo de 2-4 meses, se consigue «restablecimiento del flujo biliar». El único dato precoz que se relaciona con la probabilidad de tener buen restablecimiento de flujo es la forma anatómica de atresia extrahepática: los niños que tienen la parte proximal al hilio permeable consiguen flujo en el 100% de los casos. En contraste, los que tienen la parte distal de la vía permeable lo logran en un 63% y los afectos de atresia de toda la vía extrahepática en un 40% de los casos.
El descenso franco de la ictericia y su desaparición posterior es el parámetro más indicativo de su capacidad de supervivencia. En diferentes centros de referencia se consiguen porcentajes similares, en torno al 50% de los casos, con buen restablecimiento de flujo. Un 20% adicional de niños tiene un flujo biliar escaso, lo que modifica sólo ligeramente el mal pronóstico natural de la enfermedad. Los niños que no restablecen el flujo biliar, una vez operados por un equipo habituado a esta afección, se cree representan pacientes que tenían ya ocluidos los conductos biliares cercanos al hilio, o bien una enfermedad hepática más grave basal o agravada por colangitis postoperatoria que les impide producir bilis, aunque anatómicamente se haya restablecido la posibilidad de su paso a intestino.
La indicación de trasplante se basa en la historia natural de la enfermedad, bien conocida por su frecuencia y bastante uniformidad entre pacientes. La mortalidad en el primer año en los niños no operados y en los operados con flujo biliar ausente o escaso es tan elevada que hace necesario iniciar los estudios y proponer trasplante hepático en cuanto haya habido un tiempo juicioso (2 meses) de seguimiento tras la técnica de Kasai. Ese tiempo permite descartar una posible interferencia en la valoración debida a una colangitis superpuesta. El retraso en la indicación solamente conlleva riesgos de mortalidad para el paciente, y que el trasplante se realice en peor condición clínica y nutricional. Además, en esta enfermedad existe un proceso de hipoplasia portal que en algunos puede condicionar dificultades técnicas y trombosis portal o estenosis significativa después de realizar el trasplante.
Los niños de evolución anictérica son también pacientes con lesiones hepáticas histológicas severas. Sin embargo, la mayoría tolera períodos largos de supervivencia con buena calidad de vida, sin signos analíticos de insuficiencia, aunque van progresando lentamente los indicadores de hipertensión portal, con esplenomegalia y datos de hiperesplenismo3. La curva de supervivencia con hígado propio baja bruscamente entre los 10 y 15 años debido a que en esa edad es cuando comienza a incidir la hemorragia digestiva y/o aparecen datos analíticos de insuficiencia hepática. La confirmación de esos hechos se considera un buen parámetro para indicar el trasplante. Algunos pacientes podrán sobrevivir períodos largos si no se realiza, pero si se contrasta la calidad de vida y la probabilidad de supervivencia con trasplante, es más seguro realizarlo que esperar a una descompensación final de la enfermedad. En menos de un 5% de los casos de ABE con supervivencia larga puede desarrollarse una complicación que indica trasplante, independientemente del resto de los parámetros de la funcionalidad hepática: la hipoxemia debida a vasodilatación arteriolar pulmonar y/o shunts arteriovenosos intrapulmonares.
La supervivencia hasta la edad de adulto joven es excepcional sin trasplante en pacientes con atresia biliar. No obstante, ello sucede en un 50% de los casos que tienen una parte permeable, proximal o distal, de la vía biliar cuando son operados.
Síndrome de Alagille
La enfermedad es autosómica dominante o esporádica, debida a una mutación o deleción en el gen JAG1 del cromosoma 204, que determinará un trastorno en la diferenciación de los tejidos fetales. El diagnóstico de síndrome de Alagille (SA) se basa en la existencia de al menos 3 de los siguientes rasgos mayores: facies peculiar, colestasis intrahepática con escasez ductal, cardiopatía (estenosis pulmonares periféricas, tetralogía de Fallot), defectos del cierre del arco posterior vertebral (vértebras en mariposa) y alteraciones oculares (embriotoxon posterior). Los niños pueden presentar además otros rasgos como malformaciones renales (hipoplasia, duplicidad), alteraciones genitales (criptorquidia), retraso mental, otitis recurrentes o voz atiplada5.
La forma de comienzo de la enfermedad hepática suele ser muy temprana, como ictericia colestática en el lactante. También puede ocurrir en niños de más edad, con prurito, sin ictericia como síntoma que motiva la consulta médica. La hepatopatía puede detectarse de forma incidental en los que se diagnostica antes de otra manifestación renal o cardíaca del síndrome.
El pronóstico vital está influido notablemente por las manifestaciones iniciales6. Los niños con ictericia neonatal suelen desarrollar síntomas colestáticos crónicos con prurito rebelde, xantomas y dificultades en el crecimiento7. La indicación de trasplante en SA es obvia cuando el niño evoluciona hacia signos clínicos de hipertensión portal o insuficiencia hepática. Entre los datos analíticos relacionados con progresión a insuficiencia hepática en este síndrome se incluye el descenso de la hipercolesterolemia, cuando no va acompañado de mejoría en el prurito y la ictericia.
Sin embargo, en la mayoría de los pacientes se considera y realiza el trasplante antes si la colestasis causa mala calidad de vida, hay dificultades en la relación social y el aprendizaje y retraso severo en el crecimiento a pesar de soporte nutricional. En nuestra experiencia, considerando que el trasplante se realiza en más de la mitad de los casos para aliviar los síntomas y no porque el niño presente un riesgo vital próximo, la supervivencia actuarial global con hígado propio en el SA que se manifiesta con ictericia neonatal es del 60% a los 5 años, el 36% a los 10 años y el 20% a los 18 años. En la supervivencia han influido también las lesiones cardíacas y renales asociadas, pero la hepatopatía es el principal factor. El trasplante es necesario más tempranamente en un subgrupo de niños que mediante laparotomía se les diagnosticó de atresia extrahepática, siempre proximal, en el período de lactante pequeño. En estos pacientes se interpreta como un grado superior de afectación de la vía biliar intrahepática que incluye el hilio y zona proximal de la vía extrahepática. La realización de una portoenteroanastomosis no cambia el pronóstico. El curso no es tan grave como en la ABE pero estos niños requieren trasplante entre los 2 y 5 años.
La afectación típica cardíaca, de estenosis pulmonares periféricas, no supone habitualmente contraindicación para el trasplante. Según nuestra experiencia, puede efectuarse cirugía o angioplastia percutánea para las estenosis cercanas a la válvula pulmonar; según la experiencia en Bicêtre, el trasplante pudo realizarse en niños con presión en ventrículo derecho de hasta 50% de la presión sistémica. A pesar de la notable hipercolesterolemia no hay consecuencias cardiovasculares apreciables.
La enfermedad renal merece atención; la hipoplasia renal puede no apreciarse en el niño pequeño y ser evidente años después. Respecto de la alteración psíquica, con cociente intelectual bajo, que puede formar parte de este síndrome, es difícil deslindar de la producida por la falta de descanso y la molestia permanente del prurito. Es más prudente no considerarla un obstáculo para el trasplante, pues tras el trasplante es común una mejoría espectacular en el carácter, la atención y la capacidad intelectual de estos niños.
El curso de la enfermedad hepática es totalmente distinto en los casos cuya hepatopatía no causa ictericia neonatal. En nuestro centro ningún paciente ha fallecido o necesitado trasplante entre los que fueron detectados sin ese antecedente.
Colestasis intrahepática familiar progresiva
Éste es un grupo heterogéneo de enfermedades colestáticas de la infancia, de herencia autosómica recesiva. Recientemente se han identificado en algunos pacientes defectos en funciones canaliculares: de una ATPasa tipo P implicada en la translocación de aminofosfolípidos que constituyen la bicapa lipídica en la membrana canalicular (colestasis intrahepática familiar progresiva [CIFP] tipo 1), en la bomba de transporte de sales biliares o BSEP (CIFP tipo 2) o en la secreción biliar de fosfolípidos por una alteración (carencia o degradación acelerada) en el transportador MDR3 (CIFP tipo 3)8,9. En la práctica clínica actual no es habitual disponer de estudios de secuenciación de los genes implicados en la CIFP para un diagnóstico exacto. Desde hace poco tiempo se aplican técnicas de inmunohistoquímica que informan de la expresión de los transportadores BESEP, MDR3 y MRP2.
En la mayoría de los casos, el inicio clínico de la enfermedad sucede en la etapa neonatal o de lactante, con ictericia colestática y prurito intenso. La histología inicial corresponde a una «hepatitis a células gigantes» en el caso de niños con CIFP con gammaglutamil transpeptidasa (GGT) normal, y semeja a la de una ABE, con fibrosis portal y proliferación ductal, en el caso de CIFP tipo 3.
En 23 niños con CIFP con GGT normal seguidos en nuestro centro, la inmunohistoquímica identificó 10 casos con ausencia de expresión canalicular del BSEP (CIFP tipo 2), y 12 casos con expresión normal de BSEP (probable CIFP tipo 1). Los niños BSEP negativos tienen transaminasas más elevadas, el 70% presentó algún período de remisión de la ictericia, la biopsia mostró transformación gigante celular (TGC) en el 80% de los casos; la talla se afectó en el 40%, el 60% desarrolló litiasis biliar y el 50%, descompensación grave de la hepatopatía.
Los niños BSEP positivos tenían transaminasas poco elevadas, ictericia continuada, biopsia inicial con TGC o conductos biliares pequeños. El crecimiento se afectó gravemente en el 82%, el 9% tuvo litiasis biliar y el 27% presentó descompensación hepática. Los niños con CIFP tipo 1 presentan diarrea postrasplante a veces de difícil manejo.
Actualmente el trasplante hepático está indicado si existe grave retraso estatural con colestasis causante de mala calidad de vida (por prurito asociado) refractaria a tratamiento médico con ursodeoxicólico y soporte nutricional. La realización de una derivación parcial externa de bilis o de una exclusión parcial yeyunal no está apoyada de manera uniforme, a pesar de que se han descrito casos de pacientes (9 de 16 en la primera descripción de Whitington en 1994) que experimentan un franco descenso o desaparición del prurito y de la disfunción hepática después de esos procedimientos. La heterogeneidad de la enfermedad es posible entre niños afectos de CIFP en diferentes países, lo que hace difícil extrapolar los resultados de una terapia según lo obtenido en un grupo concreto. Si se decide probar la eficacia de una derivación externa parcial de bilis es necesario hacerlo antes de que la enfermedad esté evolucionada. Solamente un 30% de los niños sobreviven con el hígado propio a los 7 años de edad. La mayoría de los niños fueron trasplantados con el criterio de tratar síntomas colestáticos intolerables; sin embargo, el hígado explantado tuvo en todos ellos una fibrosis marcada.
Hay además formas clínicas de clínica y pronóstico «intermedio» entre los CIFP y colestasis benigna recurrente. Se trata de niños con síntomas continuos pero sin fibrosis apreciable en la biopsia hepática durante largos períodos. En la figura 1 se presentan los que han tenido supervivencia más larga. El pronóstico a largo plazo no es benigno. En nuestra experiencia ha aparecido descompensación grave entre los 13 y 16 años de edad en 3 casos. Otros 2 pacientes son ahora mujeres adultas, una de ellas ha evolucionado con hígado nodular y otra tuvo un deterioro importante durante una gestación, que revirtió tras el parto.

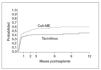
Fig. 1. Probabilidad de presentar rechazo según la inmunodepresión primaria (ciclosporina en microemulsión, n = 90 niños; tacrolimus, n = 91 niños). El primer mes es el momento de máxima incidencia de rechazo. La probabilidad acumulada a lo largo del primer año postrasplante fue de 59,8% (CSA-ME) y 44,5% (Tac) (p = 0,028). Tomada de Kelly DA, Jara P, Rodeck B, Lykavieris P, Burdelski M, Becker M, et al. Tacrolimus and steroids versus Ciclosporin microemulsion, steroids and Azathioprine in children undergoing liver transplantation: randomised European multicentre trial. Lancet. 2004;364:1054-61.
La mayor dificultad para la indicación de trasplante en CIFP con GGT normal, antes de que ocurra el deterioro con riesgo vital, se debe a la posibilidad de que en el curso postrasplante haya diarrea crónica y esteatosis hepática. En la CIFP1 el producto del gen mutado (FIC1) se expresa también en intestino delgado y páncreas. Recientemente se han reportado casos observados en el hospital de niños de Bicêtre con CIFP afectos antes del trasplante de diarrea crónica y esteatosis hepática. La mayoría de los centros de trasplante, sin embargo, ha observado ese problema en niños ya trasplantados. El restablecimiento de excreción de sales biliares postrasplante es el desencadenante de la diarrea, que cede si alguna incidencia disminuye el flujo de bilis del injerto. Esta complicación puede ser previsible si el diagnóstico pretrasplante se confirma como CIFP1, y desde nuestro punto de vista justificaría el retraso en la indicación de trasplante o el intento terapéutico de derivación parcial externa de la bilis.
La CIFP con GGT elevada (CIFP3) tiene un pronóstico variable y también menos conocido por ser menos frecuente10. En los niños el inicio es temprano, en el lactante pequeño, con ictericia y signos semejantes a una ABE. El curso posterior es hacia hipertensión portal progresiva y sus complicaciones, hechos que hacen necesario el trasplante hepático. Sin embargo, en adultos puede diagnosticarse esta entidad en forma más benigna. Es una causa de colestasis clínica durante la gestación.
Deficiencia de alfa 1 antitripsina (alfa1-AT)
La alfa1-AT se codifica en el cromosoma 14 (14q31-32). Mutaciones en ese gen condicionan la síntesis de alfa1-AT anómala. La enfermedad hepática aparece en un 10% de los nacidos con genotipo ZZ que son vigilados hasta la edad de adulto joven11,12. El cambio en un aminoácido conduce a una polimerización anormal de la proteína en el retículo endoplásmico con 2 consecuencias: su retención en el retículo endoplásmico y una escasa salida a plasma con disminución de la concentración sérica. La hepatopatía se atribuye no sólo a la retención de alfa1-AT intrahepatocitaria, hecho que sucede en todos los deficitarios, sino a la asociación de un defecto de su degradación en el retículo endoplásmico.
El 70% de los casos con hepatopatía por deficiencia de alfa1-AT presenta manifestaciones de ictericia colestática en el período neonatal. Con este antecedente, el 70% precisará trasplante hepático antes de la edad adulta. Los niños con ictericia persistente a los 6 meses de edad son el grupo de mayor riesgo de mortalidad temprana; en ellos, el trasplante es necesario antes de los 3-5 años de edad. En los demás casos la indicación de trasplante se realiza ante hipertensión portal avanzada o signos analíticos de incipiente insuficiencia13.
Otras hepatopatías metabólicas
El trasplante hepático está indicado para enfermedades hepáticas de base genética debidas a defectos enzimáticos o en sistemas de transporte que originan cirrosis. La tirosinemia I puede responder espectacularmente al tratamiento con NTBC; el trasplante está indicado en niños con enfermedad hepática aguda si no existe respuesta a NTBC en 6 semanas o si se diagnostica cuando existe ya hepatocarcinoma14. En la enfermedad de Wilson15 el trasplante es necesario si el diagnóstico se ha producido en un contexto clínico «agudo» de insuficiencia hepática con hemólisis, o en fase de hepatopatía descompensada con complicaciones que comprometen la vida a corto plazo; en caso contrario, puede restablecerse la normalidad funcional con penicilamina o trientina. El trasplante también está indicado en pacientes con cirrosis debida a fibrosis quística cuando origina hipertensión portal complicada y/o malnutrición16.
El trasplante hepático puede ser necesario en el tratamiento de enfermedades en las que existe un defecto metabólico hepático aislado, que no ocasiona compromiso de la función hepática general ni cirrosis pero determina una afectación severa de otros órganos que la sustitución hepática previene. Dentro de este grupo se incluye la enfermedad de Crigler-Najjar, la enfermedad de jarabe de arce clásico, las enfermedades del ciclo de la urea, hiperoxaluria primaria e hipercolesterolemia familiar homocigota.
Insuficiencia hepática aguda grave
La insuficiencia hepática aguda grave debida a virus, idiopática, tóxica o autoinmune tiene una elevadísima mortalidad. La posibilidad de supervivencia es mayor con trasplante, por lo que éste está indicado en todos los casos que cumplan criterios estrictos de fallo agudo (coagulopatía grave con actividad de protrombina inferior al 50% asociada a encefalopatía hepática, en un paciente sin hepatopatía previa) y que, con medidas de sostén, no muestren evolución a mejoría a corto plazo. Hay que considerar si existen factores precipitantes o agravantes que puedan ser susceptibles de tratamiento médico y cambien la apreciación de gravedad17. Ello incluye un manejo inadecuado previo, con abuso en el aporte de proteínas, diuréticos o sedantes, o la coexistencia de alteraciones iónicas o infección bacteriana.
Se han propuesto varios esquemas para indicar el trasplante en el fallo agudo, pero realmente son la descripción de este diagnóstico: coagulación severamente alterada, bilirrubina elevada y encefalopatía. A pesar de su gravedad, puede ser reversible en un pequeño porcentaje de niños, que sin embargo y lamentablemente no pueden ser identificados con seguridad para evitar su trasplante. Desde un punto de vista teórico, los pacientes de curso más rápido y grave aparente, con encefalopatía aparecida en la primera semana tras el inicio de la ictericia, son los que tienen más probabilidades de sobrevivir sin trasplante. No obstante, en niños menores de 10 años afectos de hepatitis fulminante se considera una probabilidad global de sobrevivir de un 10%. El trasplante hepático en el fallo agudo tiene un 50-70% de probabilidad de supervivencia.
Una forma de combinar la posibilidad de regeneración hepática y mejorar la supervivencia es realizar un trasplante auxiliar, un trasplante hepático en el que se conserva un lóbulo del hígado propio (derecho en el caso de fallo hiperagudo e izquierdo en caso de fallo subagudo). En el caso de que ocurra regeneración, el niño podrá volver a una vida sin inmunodepresión. Esta técnica es compleja y poco extendida, pero con resultados alentadores según la experiencia inglesa. Otros procedimientos de sostén se indican durante la espera hasta el trasplante, incluido ampliar el período de observación de una posible reversión, como la aplicación de diálisis con albúmina y el trasplante de hepatocitos.
La etiología de la enfermedad tiene importancia pues la supervivencia es mejor en el caso de hepatitis por parvovirus B19, hepatitis A y hepatitis autoinmune fulminante. Es posible que diferentes apreciaciones en la supervivencia sin trasplante se deban a la diversidad de situaciones que diferentes autores describen como «fallo hepático». Así, algunas casuísticas incluyen a niños que nunca desarrollaron encefalopatía y que por tanto no deberían considerarse en ese grupo. Por otra parte, el síndrome de fallo hepático agudo comienza generalmente con una afectación grave del hígado que sólo días o semanas después se acompañará de encefalopatía. El niño puede también fallecer a consecuencia de una hemorragia aunque no exista encefalopatía. Debe recordarse que la aparición de la encefalopatía es necesaria para aplicar el cálculo de riesgos vitales frente al trasplante, y que es muy distinto y mejor el pronóstico de los niños que no tienen esa complicación. En Francia se realizó un estudio de factores asociados al pronóstico en el fallo por hepatitis A y encontraron que un buen marcador de supervivencia fue el valor de factor VII superior al 10%.
En el caso de hepatitis autoinmune debe intentarse antes el tratamiento con esteroides, ciclosporina y azatioprina. Se desconoce el porcentaje de recuperación con estas medidas, ya que esta forma de presentación de la hepatitis autoinmune es rara. Prácticamente todos los casos son hepatitis LKM positiva en niños menores de 2 años. El intento terapéutico puede observarse unos días. En nuestra experiencia, el curso fulminante de esta enfermedad es menos rápido que en las virales y permitió seguir la evolución de los pacientes entre 10 y 15 días. En nuestro centro, el 50% de los pacientes afectados de esta forma de hepatitis autoinmune sobrevivió sin trasplante.
Tumores hepáticos
El hepatoblastoma irresecable (por afectar el segmento 4 o ambos lóbulos) y otros tumores malignos menos frecuentes no resecables y limitados en su extensión al hígado son susceptibles de trasplante. Los hemangiomas y
hemangioendoteliomas que no responden a otros tratamientos son también susceptibles de trasplante. No puede descartarse que en el seguimiento existan complicaciones graves, angiomas no detectados previamente en órganos diferentes. En los casos de hemangioendotelioma hepático hay posibilidad de recaída después del trasplante, como una neoplute;n de trasplante debida a hepatopatías crónicas virales (virus de las hepatitis B [VHB] y C [VHC]), hepatitis autoinmunitaria, colangitis esclerosante, síndrome de Budd-Chiari o hepatopatía idiopática. Estos diagnósticos constituyen menos del 10% del total de indicaciones de trasplante en series pediátricas.
CONTRAINDICACIONES DEL TRASPLANTE HEPÁTICO EN NIÑOS
Existen muy pocas condiciones que impidan la realización de trasplante en niños de cualquier edad. La presencia de una enfermedad infecciosa grave no controlada, infección sintomática por el virus de la inmunodeficiencia humana (VIH), neoplasia extrahepática y afectación neurológica significativa irreversible son las únicas contraindicaciones para el procedimiento. La posibilidad de trasplante combinado de otros órganos permite el tratamiento en niños con afecciones severas renales, intestinales, cardíacas o pulmonares.
Es importante realizar un diagnóstico correcto de la hepatopatía pues algunas enfermedades sistémicas tienen manifestaciones iniciales solamente hepáticas en las que el trasplante hepático no estaría indicado, ya que cursan con participación neurológica (como las enfermedades de la cadena respiratoria mitocondrial, enfermedad de Niemann Pick A y C, Wolman, glucogenosis tipo IV) y otras que se asocian a una recidiva de la hepatopatía prácticamente universal (como la hepatitis de células gigantes con prueba de Coombs positiva).
ESTUDIO Y PREPARACIÓN DEL CANDIDATO A TRASPLANTE
El estudio del niño al que se ha decidido someter a un trasplante incluye la «revaluación de la historia» para confirmar la etiología y los datos clínicos sobre los que se basa la indicación; se debe tomar medidas terapéuticas que permitan sostener el estado nutricional y general del paciente. Todas las hepatopatías crónicas son susceptibles de desarrollar hepatocarcinoma por lo que se aconseja la medición de la alfafetoproteína. Una valoración de los vasos mediante ecografía, y si es necesario con resonancia magnética, permite el diseño correcto de la cirugía en pacientes que tienen hipoplasia portal o trombosis grave, o en el caso de síndrome Budd-Chiari. Puede así preverse la necesidad de injertos vasculares que deben obtenerse del donante, la longitud de la cava del donante a resecar e incluso la posible necesidad de circulación extracorpórea durante el trasplante para anastomosar la cava en su porción superior.
El curso postrasplante es muy complejo al principio y es conveniente disponer antes del trasplante de una evaluación de todos los órganos vitales que permita tener una referencia. El estudio de la morfología renal y la función glomerular y tubular puede detectar hipoplasia renal, duplicidades, riñón en herradura, glomerulopatías, tubulopatías y nefrocalcinosis relacionadas con la enfermedad primaria (p. ej., en el SA), secundarias a una hepatopatía evolucionada (por raquitismo, hiperlipemia y tratamientos administrados), o asociadas fortuitamente.
La realización de una radiografía de tórax, un electrocardiograma, una ecografía cardíaca y la medición de la saturación de O2 permite excluir enfermedades, confirmar la buena función ventricular o conocer la presencia de problemas relacionados con la hepatopatía (como hipertrofia del ventrículo derecho y shunts arteriovenosos pulmonares). Es conveniente realizar un electroencefalograma en todos los niños, pues proporciona una referencia en el caso de que ocurran crisis convulsivas postrasplante.
El cribado de enfermedades infecciosas obliga a realizar Mantoux, serología frente a VHB, VHC, virus de Epstein-Barr (VEB), VIH, citomegalovirus, herpes simple y toxoplasma. Tiene que confirmarse la protección del niño frente a VHB y VHA tras recibir la vacunación. Está bien documentada la aparición de hepatitis B de novo en pacientes no protegidos que reciben un injerto hepático de un donante HBsAg negativo pero anti-HBc positivo. El niño candidato a trasplante debería estar siempre ya vacunado frente a difteria, tétanos y tos ferina, polio, hemofilus, neumococo, meningococo, hepatitis B, hepatitis A y, si el tiempo lo ha permitido, frente a sarampión, rubéola y parotiditis. Las vacunas de virus vivos no deben administrarse en el plazo de 1 mes anterior al trasplante.
No es precisa la determinación de HLA pues no se usará para seleccionar el donante. La compatibilidad del grupo sanguíneo ABO es el único parámetro.
Según los antecedentes del paciente puede ser necesario completar la evaluación con un estudio de enzimas eritrocitarias (antecedente de anemización con fármacos), de función plaquetaria (en todos los tipos de glucogenosis y en las hepatopatías idiopáticas), descartar una deficiencia de inmunoglobulina A y hacer una prueba de Coombs (en caso de enfermedad de base autoinmunitaria), realizar pruebas de función pulmonar (fibrosis quística, deficiencia de alfa1-AT) o una búsqueda en todos los órganos para detectar angiomas (hemangiomas o hemangioendoteliomas).
LA CIRUGÍA DE TRASPLANTE HEPÁTICO INFANTIL
La técnica clásica de trasplante consiste en la extracción del hígado enfermo con la cava retrohepática, seguida del implante de un injerto completo. La reconstrucción se realiza con la anastomosis de la cava inferior del donante en sus 2 extremos, superior e inferior, a la del receptor. La revascularización portal se efectúa mediante una anastomosis terminoterminal entre la vena porta del injerto y la del receptor. La revascularización arterial suele efectuarse con la boca confeccionada con la arteria gastroduodenal y la arteria hepática del receptor, y el tronco celíaco del donante18. La reconstrucción biliar en niños es casi siempre una hepaticoyeyunostomía en Y de Roux.
Alrededor del 40% de los niños que deben someterse a trasplante hepático son menores de 2 años de edad19. La técnica de trasplante con injerto entero precisa que el donante tenga similar edad, y ello limita profundamente la accesibilidad. Desde 1985 se practica una técnica alternativa que permite, mediante la reducción del injerto, adecuar el tamaño al del receptor de donantes que le superan hasta 6 veces en peso. Entre 1995 y 2000, el 54% de los trasplantes en niños en Estados Unidos se realizó con injerto reducido; en nuestro centro el porcentaje en los últimos 5 años fue del 56%19,20.
El injerto reducido se realiza en el lóbulo izquierdo (con resección habitual del segmento I) o en el segmento lateral izquierdo (segmentos II y III). La partición tiene lugar en el banco (ex situ). La cava del receptor debe conservarse resecándola del hígado enfermo, para evitar una excesiva desproporción en el calibre con el donante. La vena suprahepática del injerto se anastomosa terminolateral en la cava inferior del receptor.
En el trasplante de injerto reducido puede, en lugar de despreciar el resto del hígado, utilizarse el lóbulo D para el trasplante en un niño mayor o un adulto (técnica split; fig. 2). Es una técnica más compleja pues deben mantenerse viables ambos lóbulos y sus pedículos vasculares.
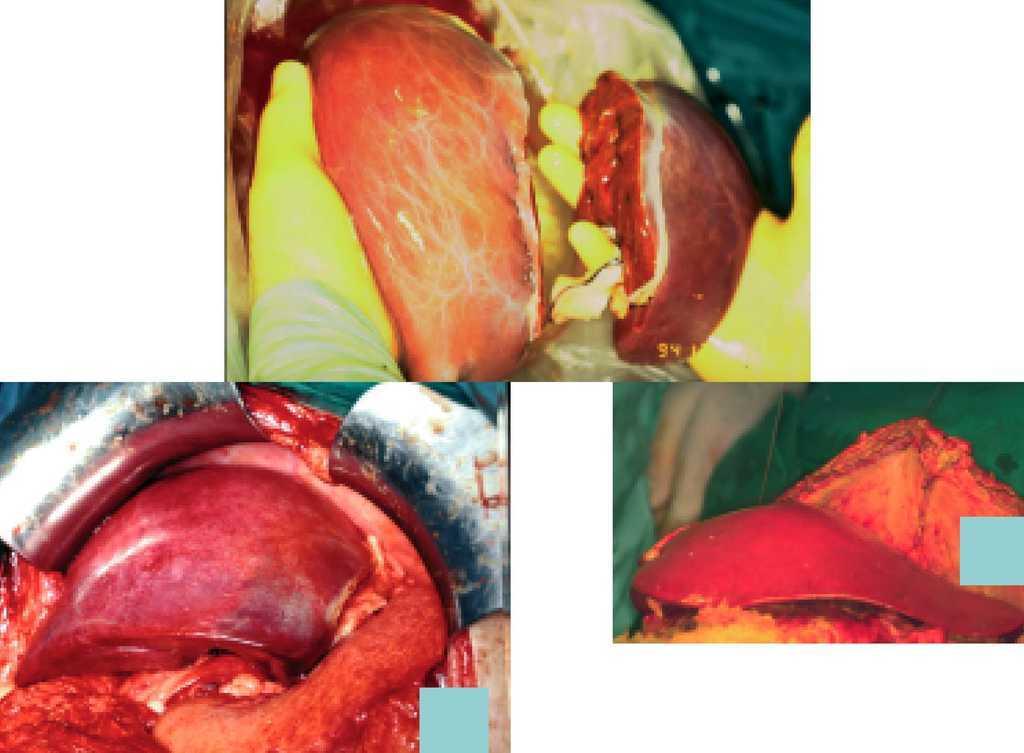
Fig. 2. Técnica quirúrgica del trasplante hepático en niños.
Desde 1995, la técnica de trasplante con donante vivo para niños, con el empleo del segmento lateral izquierdo del donante, es una práctica en auge21-23. Frente a un injerto con técnica similar (split) procedente de cadáver, la función inicial es mejor en el injerto de donante vivo, aunque la evolución posterior no presenta diferencias significativas24-27.
Aunque los resultados finales de supervivencia con técnicas de trasplante reducido son semejantes a los obtenidos con injerto entero, la morbilidad es mayor dado que la partición repercute más en la función inicial del injerto, aumenta la tasa de fallo primario, ocasiona más riesgo de hemorragia postoperatoria por la zona de corte y tiene una incidencia mayor de fugas y estenosis biliar, o dificultades en el flujo de las venas suprahepáticas debido a la rotación que presenta el hígado para adaptarse en el espacio subdiafragmático. Estos problemas se consideran aceptables porque a cambio de ello es posible el trasplante en un plazo adecuado. En los niños pequeños el trasplante reducido ofrece una ventaja significativa frente al trasplante de injerto entero ya que disminuye el riesgo de trombosis arterial.
La reconstrucción biliar en niños se realiza comúnmente mediante hepaticoyeyunostomía28. Esta reconstrucción tiene más riesgo de favorecer una colangitis ascendente. La reconstrucción hepática y del colédoco tiene una tasa cercana al 50% de estenosis y fístula, por lo que se reserva para niños mayores con vía biliar no afecta por la enfermedad original. El trasplante dominó ha sido factible en algunas enfermedades metabólicas29.
Muchas complicaciones postrasplante guardan relación con los datos de la cirugía. Los tiempos quirúrgicos, como la duración total, del período anhepático, de la isquemia fría y los volúmenes de transfusión de hemoderivados son buenos indicadores, si sobrepasan la media, de problemas diversos que tienen repercusiones en el post-
operatorio inmediato: disección difícil por adherencias, perforaciones intestinales durante el procedimiento, dificultad en alguna de las anastomosis con reconstrucción subóptima o que precisa injertos interpuestos. La impresión buena del injerto una vez reperfundido y la ausencia de hemorragia importante serán datos muy relacionados con el buen pronóstico.
LA INMUNOdePRESIÓN
El rechazo del injerto está mediado por la reacción de las células T del receptor frente a los antígenos alogénicos expresados en el injerto. El reconocimiento de esos antígenos pone en marcha la liberación de interleucina 1 (IL-1) e IL-2, con la consiguiente activación y proliferación de células CD4 y CD8. Se produce luego una gran variedad de citocinas que estimulan el reclutamiento de linfocitos hacia el espacio porta y modulan la expresión de moléculas de adhesión.
El tratamiento inmunodepresor desde el implante es la clave para conseguir la supervivencia del injerto y del paciente. Solamente la disponibilidad de un fármaco potente, la ciclosporina, en 1978, pudo conseguir el éxito de los trasplantes y la proliferación de esta técnica en todo el mundo. Otro fármaco inmunodepresor, el tacrolimus, marcó desde su introducción en 1989 un gran avance para la supervivencia.
El diseño de la inmunodepresión ha tenido grandes diferencias entre centros y países30. Actualmente es más uniforme. Los pilares fundamentales son los fármacos de acción inhibidora de la calcineurina (ciclosporina o tacrolimus). Los esteroides se asocian inicialmente. Sin embargo, persisten diferencias entre centros en cuanto a los valores deseados de los fármacos en los diferentes tiempos postrasplante, en la dosis del esteroide, la intención de retirada de esteroides y el momento de ésta31. La incorporación de nuevos agentes como la rapamicina (sirolimus)32,33, el micofenolato mofetil34 y los anticuerpos antirreceptor de IL-2 (basiliximab o daclizumab)35 ofrece opciones alternativas (asociados a un anticalcineurínico y esteroide) de inmunodepresión primaria en pacientes de riesgo alto, o sustituye a los inhibidores de calcineurina en pacientes con insuficiencia renal o si hay signos de toxicidad grave con los fármacos convencionales.
La búsqueda de un balance adecuado entre la inmunodepresión y sus riesgos parece concluir hacia la conveniencia de una inmunodepresión potente en el postoperatorio inmediato (inducción), y proteger al paciente de infección bacteriana, fúngica y viral en ese período. Es a un plazo medio y largo cuando con el conocimiento de su comportamiento previo se da más importancia a la individualización y búsqueda de la inmunodepresión mínima eficaz.
Los fármacos inmunodepresores principales
Tacrolimus
Es un antibiótico macrólido aislado del hongo Streptomyces tsukubaensis. Tiene una proteína ligadora intracelular en el linfocito (FKBP-12). La formación de un complejo de tacrolimus-FKBP-12, calcio, calmodulina y calcineurina causa una inhibición de la actividad fosfatasa de la calcineurina. Ello impide la generación del factor nuclear de las células T activadas, una proteína que inicia la transcripción de los genes de citocinas. La producción de IL-2 se inhibe y, en consecuencia, la proliferación de células T, la expansión clonal y la producción de citocinas.
La dosis inicial en niños es de 0,3 mg/kg/día (0,15 mg/kg/día en mayores) cada 12 h, por sonda nasogástrica. Los valores en valle se correlacionan bien con el área bajo la curva de concentración-tiempo (AUC). Con la medición del valor en sangre (diaria en la primera semana) se corrige en cada individuo la dosis para lograr las concentraciones deseadas. La absorción intestinal es variable, entre individuos, en las diferentes fases de evolución en el postrasplante inmediato, y disminuye si coincide la ingesta de alimentos, pero es poco dependiente de la presencia de sales biliares. La eliminación ocurre por metabolización hepática a través del sistema de citocromo P450. Menos del 1% se excreta en orina.
Ciclosporina en microemulsión
La ciclosporina es un endecapéptido cíclico obtenido del hongo Tolipocladium inflatum. Tiene un receptor intracelular denominado ciclofilina. El complejo ciclosporina-
ciclofilina se une a calcineurina e inhibe su actividad fosfatasa. El mecanismo inmunodepresor es por tanto el mismo que en el caso del tacrolimus. La mayor potencia del tacrolimus (por 10 in vivo, por 100 in vitro) se debe a la mayor afinidad de su complejo con FKBP12 por la calcineurina.
La formulación de ciclosporina en microemulsión usa una mezcla de surfactante y solventes lipofílicos e hidrofílicos que permite una mejor absorción del fármaco, menos dependiente de las sales biliares que la ciclosporina original. Su desarrollo permitió el uso de este fármaco por sonda nasogástrica desde las primeras horas postrasplante. La dosis inicial es de 15 mg/kg/día (10 mg/kg/día en mayores) repartido en 2 tomas. La medición de la concentración en sangre permite los ajustes de dosis correspondientes, ya que existe una importante variabilidad en la absorción entre individuos y, como en el caso del tacrolimus, la eliminación se realiza por metabolización por parte de citocromo P450, y por lo tanto se modifica según la función del injerto. Los valores en valle no tienen buena correlación con el AUC. Actualmente hay suficiente evidencia para sustituir el tradicional control mediante valores en valle por el control basado en el valor a las 2 h
postadministración, que muestra una mejor correlación con el AUC36,37. No obstante, aún no hay una amplia experiencia en cuanto a los valores de C2 deseables en el postoperatorio inicial y tardío. En la incipiente práctica en nuestro centro observamos, en pacientes a largo plazo postrasplante, mucha variabilidad de C2 con la misma dosis en controles sucesivos de un mismo individuo.
Modelos básicos de inmunodepresión postrasplante
Los modelos vigentes para inmunodepresión primaria consisten en el uso de tacrolimus más esteroides, o de ciclosporina en microemulsión más esteroides; esta última pauta se complementa en los primeros meses con azatioprina. La pauta basada en tacrolimus resulta muy adecuada para niños y jóvenes ya que no causa hiperplasia gingival ni hipertricosis38. Sin embargo, las complicaciones derivadas de su gran potencia inmunodepresora pueden ser más graves que con el uso de ciclosporina. Recientemente finalizó un ensayo europeo en niños dirigido a evaluar esas 2 pautas de inmunodepresión primaria en cuanto a su eficacia y toxicidad39. Las dosis y los valores de fármaco se reflejan en la figura 3. Ambas pautas demostraron una elevada eficacia. La probabilidad acumulada de rechazo en el primer año fue ligeramente más alta con ciclosporina (fig. 1). La tasa de niños afectos por rechazo refractario a esteroides fue notablemente más elevada cuando la inmunodepresión primaria se basó en ciclosporina (el 26,7 frente al 5,5% en el grupo que recibió tacrolimus). La supervivencia del injerto no se modificó entre ambas pautas, debido a que en el caso de emplear ciclosporina se efectúa su conversión por tacrolimus en los pacientes que muestran rechazo refractario al esteroide. A un año de seguimiento no hubo diferencia significativa en la función renal, la incidencia de hipertensión arterial ni de la diabetes. Sólo en el grupo tratado con tacrolimus se detectaron casos aislados de miocardiopatía hipertrófica y síndrome linfoproliferativo (PTLD) (sin diferencia significativa). Se observó una frecuencia significativamente mayor de infección por VEB en niños que recibieron tacrolimus, y una incidencia exclusiva en el grupo tratado con ciclosporina de hipertricosis (28%) e hiperplasia gingival (9%).
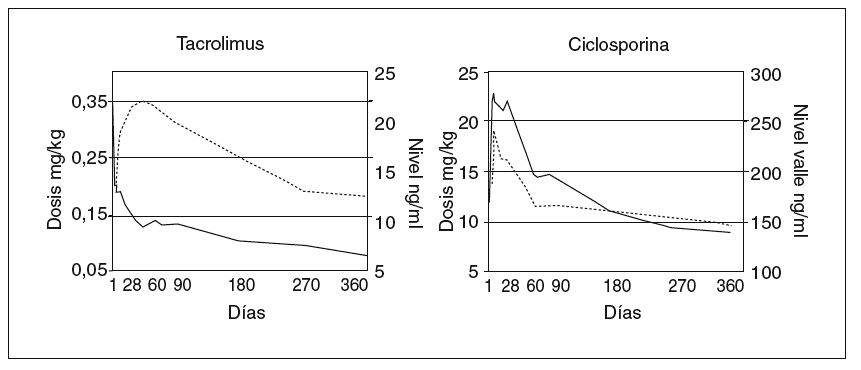
Fig. 3. Modelos de inmunodepresión primaria después de trasplante hepático. Dosis diaria vía oral y nivel en valle de tacrolimus y ciclosporina a lo largo del primer año del trasplante. Tomada de Kelly DA, Jara P, Rodeck B, Lykavieris P, Burdelski M, Becker M, et al. Tacrolimus and steroids versus Ciclosporin microemulsion, steroids and Azathioprine in children undergoing liver transplantation: randomised European multicentre trial. Lancet. 2004;364:1054-61.
COMPLICACIONES DEL TRASPLANTE HEPÁTICO
El postoperatorio inicial
La duración de la hospitalización tras el trasplante está determinada por las frecuentes complicaciones que tienen lugar tras una cirugía agresiva, por la necesidad de administrar tratamientos preventivos antivirales prolongados, y la conveniencia de haber obtenido una condición estable del paciente que le permita la independencia de un centro hospitalario.
En nuestro centro, durante el período 1997-2001, la duración de la hospitalización inicial fue como media de 43 ± 17 días en niños que recibieron un injerto entero, de 58 ± 31 días en niños con un injerto reducido, y de 72 ± 35 días en niños con injerto segmentario de donante cadáver o vivo. Una hospitalización mucho menos prolongada, de 19 días de media, se reporta en la experiencia reciente de centros norteamericanos19, lo que refleja probablemente la posibilidad de tener una estancia cercana al hospital tras el alta. En nuestro centro, con un seguimiento medio de 2 años, el 40,5% no precisó reingresos, el 24% lo necesitó en 1 ocasión, el 16% en 2 y el 19% múltiples veces. La tasa de rehospitalización es mucho mayor en la experiencia norteamericana: fue necesaria en al menos 1 ocasión en el 64,8% de los niños entre el primer y sexto mes, en el 45,2% en el segundo semestre y en el 36,5% entre 12 y 18 meses después del trasplante. Estos datos indican que el postoperatorio del trasplante es complejo inicialmente. Aun obteniendo buenos resultados el camino a la estabilización precisa vigilar y tratar problemas que pueden afectar a cualquier órgano.
Afecciones generales en los períodos temprano y tardío
La función renal
En nuestro centro un 15% de los niños tenía un filtrado glomerular (FG) inferior a 80 ml/min antes del trasplante. En el 75% de éstos se debía a un síndrome hepatorrenal, en el 12% a una nefropatía estructural y en el 12% a nefropatía tóxica por quimioterapia. La cirugía del trasplante tiene repercusión sobre la función renal, por el pinzamiento de la cava inferior durante la fase anhepática, que impide el retorno venoso renal, y por estados de hemorragia intraoperatoria o hipotensión. Además, el uso de ciclosporina o tacrolimus determina una vasoconstricción arteriolar renal40. Las manifestaciones son de oliguria y descenso de FG. El aporte de líquidos debe ajustarse a las pérdidas para evitar la retención excesiva de agua. Los diuréticos, usualmente furosemida, son necesarios en casi todos los pacientes en los primeros días de postoperatorio para compaginar las entradas obligadas de agua en medicaciones y nutrientes.
En el momento de máxima alteración de creatinina sérica en el postoperatorio inmediato, el 60% de nuestros pacientes tuvo un FG inferior a 80 ml/min (en un 32% fue menor de 60 ml/min) y un 7,7% precisó medidas de soporte con hemofiltración o hemodiafiltración. La necesidad de esta técnica se asocia a afecciones graves como fracaso del injerto o la hemorragia, lo que explica que un 50% de los niños que la precisaron fallecieran.
El seguimiento indica una recuperación renal antes del segundo mes postrasplante. A una media de 2 años después del trasplante sólo un 5% de niños tiene un FG (estimado por la creatinina sérica y la talla) inferior a 60 ml/min y el 80% tiene más de 80 ml/min. Un estudio en Suecia basado en la determinación exacta del FG con aclaramiento de inulina demostró que los niños trasplantados en conjunto tienen un descenso de FG postrasplante (de 140 ± 11 ml/min/1,73 m2 pretrasplante a 95 ± 8 al año) que desciende más a lo largo de los 6 años siguientes40. Sin embargo, en cada individuo no observaron empeoramiento entre los valores al año y al sexto año. La nefropatía a medio y largo plazo se atribuye principalmente a la terapia con anticalcineurínicos. Los datos histológicos obtenidos en autopsias muestran lesiones intersticiales con fibrosis y atrofia tubular significativas, pero no siempre asociadas a disfunción renal notable. No obstante, en nuestros pacientes la nefrotoxicidad como causa aislada solamente justifica el 30% de los casos con FG inferior a 60 ml/min; en los demás había factores muy importantes añadidos como la coexistencia de una nefropatía estructural previa o la administración pre y postrasplante de quimioterapia antineoplásica. La función renal de los niños sometidos a trasplante debido a hepatoblastoma está más afectada que en niños con cualquier otro diagnóstico pretrasplante.
A medio y largo plazo son frecuentes los problemas de función tubular atribuibles a la toxicidad de los fármacos anticalcineurínicos. El 80% de los pacientes tiene necesidad de suplementos de magnesio y de bicarbonato. La hiperpotasemia ocurre en el 20% de los niños. El tratamiento de la hiperpotasemia incluye evitar alimentos ricos en potasio, dar suplementos de bicarbonato, y si persiste, en ausencia de hipertensión arterial, administrar fludrocortisona. El uso de furosemida no es recomendable porque causa un empeoramiento en el FG debido a deshidratación.
Merece especial atención otro componente de la tubulopatía tóxica, la falta de capacidad de concentración urinaria. Se expresa en una mayor ingesta de agua y nicturia, con enuresis en muchos niños. Debido a la hipostenuria, los procesos intercurrentes banales que se acompañan de vómitos o diarrea determinan deshidratación e insuficiencia renal prerrenal, incluso en períodos de enfermedad muy cortos. El ingreso en el hospital y la administración de sueroterapia se aconsejan para evitar que pueda haber repercusiones permanentes sobre el FG.
Problemas cardiopulmonares
El derrame pleural derecho afecta a más del 70% de los pacientes en el postoperatorio inmediato. Los niños pequeños pueden presentar paresia diafragmática D por manipulación intraoperatoria, o dificultad para los movimientos respiratorios debido a un tamaño grande del injerto, lo que hace necesario prolongar el soporte ventilatorio o incluso efectuar una plicatura. El derrame pleural cuantioso puede ser expresión de una paresia de diafragma. El derrame pleural prolongado puede ser un signo de estenosis de venas suprahepáticas.
La hipertensión arterial es muy frecuente en el primer mes postrasplante, cuando en casi todos los pacientes es preciso el tratamiento a demanda de hidralazina o nifedipina. El empleo de dosis menores de esteroides y la administración oral habitual de tacrolimus o ciclosporina han disminuido la prevalencia de este problema. Sólo un 8% de los niños tiene hipertensión arterial a medio y largo plazo, habitualmente sin relación con un descenso de FG. El tratamiento más adecuado es la administración de nifedipina. Sin embargo, se ha descrito que en niños trasplantados el patrón circadiano normal con descenso nocturno de la presión arterial está abolido y por tanto no puede excluirse que a muy largo plazo sean candidatos a complicaciones derivadas de la hipertensión arterial. Durante la infancia, casos aislados (< 1%) de miocardiopatía hipertrófica pueden apreciarse en pacientes con fracaso del injerto, o en niños que reciben tacrolimus como inmunodepresor.
Infecciones
La cirugía larga debido a adherencias de cirugías previas, la realización de hepaticoenterostomía, la necesidad de una malla abdominal por un tamaño excesivo del injerto, la hemorragia digestiva, la necesidad de reoperaciones, perforaciones digestivas y la hemorragia abdominal posquirúrgica son factores de riesgo de infección bacteriana y fúngica. En los pacientes pediátricos se administra descontaminación intestinal (nistatina, tobramicina y colimicina) y una profilaxis antibiótica perioperatoria (con vancomicina y aztreonam en nuestro centro) que se continúa posteriormente según los hallazgos obtenidos en cultivos rutinarios o empíricamente según los problemas evolutivos presentados (atelectasias, fístulas, trombosis, perforación intestinal, etc.). La hemorragia postoperatoria es un factor de riesgo significativo favorecedor de infección fúngica, que a nuestro juicio indica la administración preventiva de anfotericina liposomal.
El diagnóstico cierto de una infección varía según los criterios que se apliquen. En la casuística norteamericana se consideró afectos de infección bacteriana al 42% de los niños trasplantados y el 11% tuvo infecciones fúngicas. En nuestro centro, usando criterios muy amplios (un cultivo positivo con o sin clínica) un 71% de los pacientes tuvo infección bacteriana y un 25%, fúngica19,41,42. Las infecciones otorrinolaringológicas y neumonías no parecen incidir con más frecuencia en niños trasplantados ya reintegrados a la escuela. Las colangitis pueden aparecer en pacientes que evolutivamente desarrollan estenosis biliar, y en el 20% de los casos en que la ecografía ha detectado aerobilia en los controles rutinarios. Las otitis supuradas pueden reflejar una infección por VEB causal de hipertrofia adenoidea.
La infección por Pneumocistis carinii es un riesgo que se previene muy satisfactoriamente con sulfametoxazol- trimetoprima. Hay diferentes pautas de administración. En nuestro centro iniciamos la profilaxis en la tercera semana postrasplante a dosis de 5 mg/kg/día de trimetoprima, y prosigue de forma indefinida a 3 mg/kg/día. No hemos observado ningún caso de infección por este agente.
Las infecciones por citomegalovirus y VEB en niños trasplantados son frecuentes y potencialmente graves43. La primoinfección y el grado de inmunodepresión postrasplante determinan la gravedad. Asimismo, en los pacientes previamente seropositivos la reactivación en el período postoperatorio inicial puede causar síntomas graves. En los niños menores de 2 años y en los pacientes seronegativos es fundamental la prevención, pues el injerto y los hemoderivados que reciben aseguran la trel alta hospitalaria en todos los niños es una práctica común. El ganciclovir es virostático y no impide la infección por citomegalovirus o EBV pero protege del desarrollo de síntomas en un período crucial de debilidad general e inmunodepresión intensa. La alternativa es la vigilancia (antigenemia frente al citomegalovirus o viremia frente al VEB) y tratamiento en caso de signos de infección, pero no suele adoptarse esta actitud debido a la mayor dificultad de control de la infección una vez establecida.
El riesgo de infección sintomática por citomegalovirus se previene bien con la administración de un tratamiento con ganciclovir por vía intravenosa en el período postoperatorio, que se amplía más de un mes si incide un problema debilitante del paciente (o la necesidad de una intensa inmunodepresión por rechazo), hasta que se supere.
La infección por VEB es más problemática debido a su implicación en el desarrollo de un PTLD45. Esta complicación confirma los resultados de supervivencia del injerto y del paciente que ha superado el postoperatorio precoz. Actualmente es el problema menos dominado y más heterogéneamente prevenido entre los diversos centros. Para evitar el desarrollo de un PTLD caben varias actitudes. Una es la ampliación del período de tratamiento con aciclovir o ganciclovir en el postoperatorio, hasta el tercer mes o incluso más tiempo. Esta medida se asoció a un descenso en la tasa de PTLD en la experiencia de UCLA. El control estrecho, cada 3 meses, de ADN-VEB en sangre por reacción en cadena de la polimerasa indicaría la reanudación de tratamiento con aciclovir o ganciclovir, por vía intravenosa u oral según la clínica asociada. El dominio persistente de la infección, con viremias continuadas negativas, sería el criterio de retirada de los antivirales. La inmunodepresión debe mantenerse dentro de los límites normales salvo que se observen síntomas o signos incipientes o evidentes de linfoproliferación, ante lo cual es precisa su disminución o la retirada. Al aplicar la pauta de prevención antiviral durante los 3 primeros meses y la vigilancia de VEB con tratamiento si la viremia es positiva, en nuestro centro observamos en los últimos 5 años una prevalencia de infección por VEB del 53% en niños menores de 2 años y del 23% en niños de mayor edad; el 12% de los primeros y el 4% de los segundos desarrollaron PTLD.
Otras actitudes preventivas o terapéuticas dependen de la disponibilidad de unas técnicas hasta ahora no comerciales: la cuantificación de VEB en sangre y de linfocitos sensibilizados frente al VEB. El riesgo de PTLD se relacionó con el grado de viremia que el paciente presentaba. Sin embargo, no es determinante que un valor muy elevado de viremia se siga de PTLD. El riesgo de PTLD se ha relacionado recientemente de manera más fidedigna con una baja o nula proporción de linfocitos sensibilizados contra el EBV. Si esos parámetros están disponibles permiten limitar a los niños con riesgo las medidas de tratamiento antiviral y cierta reducción de inmunodepresión que de otra forma se aplican a todos los pacientes afectos de infección por VEB activa.
Problemas del injerto
La función del injerto recién implantado se caracteriza por una elevación de transaminasas y de bilirrubina debida al sufrimiento del tejido en el proceso de extracción y conservación hasta el trasplante. La alteración máxima en los 2 primeros días es más pronunciada si el injerto ha sido partido. En nuestro centro, en injertos enteros la cifra media de aspartato aminotransferasa (AST) fue de 736 U/l, la bilirrubina media de 5,6 mg/dl, la actividad de protrombina media de 53%. En injertos reducidos los valores medios fueron AST de 1.200 U/l, bilirrubina de 8,2 mg/dl y APP de 45%. Una evolución rápida hacia mejoría tiene lugar a partir del tercer día y la normalidad funcional completa se alcanza en 15 y 7 días como media, para injertos reducidos y enteros, respectivamente.
Mala función inicial y fallo primario del injerto
Por causas generalmente poco previsibles, teniendo en cuenta los datos clínicos y analíticos disponibles del donante y el aspecto externo del hígado antes de su extracción, un porcentaje de injertos tiene alteraciones analíticas muy pronunciadas una vez implantados19,20. Son factores de riesgo, según los análisis de los trasplantes hepáticos en adultos, la hipernatremia y la esteatosis hepática en el donante. En nuestra experiencia, con implantes realizados siempre con tiempos de isquemia fría inferiores a 12 h y en ningún caso con esteatosis o necrosis marcada en la biopsia del injerto tras su revascularización, el 15% de los injertos tuvo transaminasas superiores a 3.000 U/l y actividad de protrombina inferior a 35% en las primeras horas. La evolución en las horas siguientes distingue una progreso a recuperación (mala función inicial) o a fracaso, con ictericia progresiva y coagulopatía persistente aunque comúnmente exista un descenso rápido de transaminasas (fallo primario). La frecuencia de fallo primario fue del 4% del total de trasplantes. El retrasplante es necesario de forma urgente. El hígado explantado muestra amplias áreas de necrosis lobulillar hemorrágica.
Trombosis arterial
La trombosis de la arteria hepática afecta al 7% de los injertos. La forma clínica más frecuente es el desarrollo de fallo agudo del injerto; en casos aislados se manifiesta por fístula biliar o absceso en zonas del hígado previamente infartadas. Es una complicación casi exclusiva del postoperatorio temprano y principalmente causada por dificultades técnicas en la anastomosis quirúrgica y retrasplantes, aunque sobre un vaso de flujo comprometido puede influir en la formación del trombo la hiperviscosidad sanguínea, la policitemia o el aumento de resistencia al flujo causado por rechazo u otra causa de inflamación en el injerto19,20. A lo largo de los años, las técnicas quirúrgicas han sufrido modificaciones con el fin de reducir la tasa de trombosis, y se ha llegado a la conclusión de que una reconstrucción terminoterminal es la preferible.
La prevención de trombosis, con heparina en perfusión continua de 240 U/kg/día, comienza cuando se restablecen la coagulopatía y trombopenia usuales en las primeras 24-48 h y el paciente no presenta hemorragia activa. La profilaxis se extiende el primer mes, y se sustituye la heparina por dipiridamol (o aspirina en otros centros).
La vigilancia de esta complicación es necesaria en todos los niños; se realiza mediante eco-Doppler intraoperatoria y diaria en la primera semana. Mediante ella pueden detectarse situaciones de riesgo, patrones de flujo que hacen necesaria la vigilancia más estrecha, o permite un diagnóstico temprano de la complicación. El diferente calibre de la arteria hepática entre el donante y el receptor justifica que en algunos niños se observen patrones de flujo arterial semejantes al de una estenosis que evolutivamente se revelan como fisiológicos y se resuelven.
La trombosis arterial puede tratarse con trombólisis con urocinasa local; sin embargo, esta técnica puede causar hemorragia si se aplica en los primeros días tras el trasplante. En algunos casos se logra prolongar unos días la supervivencia del injerto pero el tratamiento final fue el retrasplante en todos nuestros pacientes.
Trombosis portal
La trombosis portal afecta a niños con hipoplasia del vaso grave antes del trasplante, y a los que tienen antecedente de retrasplante. La trombosis portal es una complicación rara en el postoperatorio inmediato (1%), momento en que causa fracaso del injerto e indica retrasplante urgente. El diagnóstico temprano permite una reintervención con trombectomía que puede salvar el injerto19,20.
A pesar del seguimiento estrecho ecográfico, muchos más casos de afección portal pasan inadvertidos o bien se desarrollan en un período postoperatorio tardío. En nuestro hospital un 5 y un 2,5% de los niños tuvieron estenosis portal y trombosis, respectivamente, que solamente se detectaron en el seguimiento a medio plazo. No determinaron alteraciones de la función del injerto pero hubo complicaciones de hipertensión portal prehepática. Es posible que la buena tolerancia del injerto a esta situación se deba a que desarrollan, ayudados por las adherencias quirúrgicas y la yeyunostomía en Y de Roux, una abundante circulación colateral hacia el hígado, sorteando la obstrucción. No obstante, la morbilidad por hemorragia es importante y la solución quirúrgica difícil. Entre las técnicas disponibles se prefiere el shunt de Rex, consistente en la interposición de un injerto entre la vena mesentérica y la porta izquierda, ya que así se evita el robo de flujo portal al hígado característico de otras técnicas de shunt. La detección de estenosis portal severa o con síntomas de hipertensión portal puede tratarse con buenos resultados con una dilatación con balón por vía transhepática.
Estenosis de la vena suprahepática o cava
Según la reconstrucción quirúrgica en el trasplante, pueden ocurrir estenosis en la anastomosis de cava o en la anastomosis de suprahepática a la cava del receptor. En nuestros niños ocurrió de forma temprana en el 3% y otro 2,5% la manifestó de forma tardía. La ecografía no permite detectar adecuadamente este problema y debe realizarse una angiorresonancia o exploración vascular si hay síntomas sugerentes. El derrame pleural prolongado o cuantioso en el postoperatorio inmediato, el derrame pleural aparecido evolutivamente, el aumento de tamaño del injerto o una disfunción caracterizada por descenso de la actividad de protrombina con escasa o nula alteración en otros parámetros son datos que sugieren un síndrome de Budd-Chiari. La biopsia hepática no suele ayudar inicialmente pues no muestra los signos característicos de dilatación sinusoidal y necrosis/esclerosis pericentral hasta que el problema está muy evolucionado. El tratamiento con angioplastia percutánea es resolutivo.
Rechazo del injerto
El rechazo es la complicación más frecuente del injerto. El diagnóstico es histológico. Las lesiones en el rechazo agudo (o «celular») son clásicamente una reacción inflamatoria de los espacios porta, incluidos linfocitos blásticos, polinucleares y eosinófilos, cambios inflamatorios y displásicos en el epitelio de los conductos biliares e inflamación subendotelial de las ramas venosas portales y centrolobulillares. La severidad histológica del rechazo se gradúa actualmente siguiendo el esquema de Banff46 pero la verdadera severidad del proceso se conceptúa clínicamente en función de la respuesta a la terapéutica. El diagnóstico histológico es fácil en el primer mes. Más tarde los hallazgos histológicos pueden ser menos notables, no incluir alguno de esos componentes de lesión, o las lesiones histológicas «típicas» pueden corresponder a otras causas, como una hepatitis viral.
El rechazo crónico («ductopénico y arteriopático») se define también histológicamente por destrucción ductal con o sin proliferación intimal con xantomización en las arterias de grueso y mediano calibre. Surge en la evolución de un rechazo agudo recidivante y no respondedor a la terapia, con colestasis intensa (por ductopenia). La evolución rápida a insuficiencia hepática sucede en los pacientes con arteriopatía obliterativa asociada. El diagnóstico de confirmación suele requerir el estudio del hígado explantado. En las biopsias previas pueden haberse detectado antes lesiones diferentes, como citólisis centrolobular, o necrosis centrolobular47 hemorrágica con fibrosis de las vénulas hepáticas48,49.
El rechazo agudo afecta clínicamente, con disfunción del injerto asociada a veces a fiebre, leucocitosis con eosinofilia y afectación general, a un 45-60% de los niños. Si se realizan biopsias rutinarias aun en ausencia de síntomas, hasta un 80% de trasplantados tienen lesiones de rechazo. Surge más comúnmente entre el séptimo y vigésimo primer día. séptimo y vigésimo primer día. Según las diferentes pautas de inmunosupresión basal, el riesgo pasa a ser mínimo en el seguimiento posterior o bien puede seguir incidiendo.
Los progresos de los fármacos inmunosupresores en la última década no han modificado notablemente la incidencia de rechazo pero han influido muy favorablemente en 2 aspectos. Uno es la mejor tolerabilidad de los fármacos, que tienen menos efectos adversos neurológicos en administración oral, prácticamente siempre suficiente desde las primeras horas del postoperatorio, por sonda nasogástrica. Otro es la menor gravedad de los episodios de rechazo, sólo excepcionalmente (menos del 4% del total de trasplantes) refractarios a tratamiento médico20,39.
El tratamiento del rechazo agudo consiste en la intensificación del tratamiento inmunodepresor, con la administración de corticoides (45 mg/kg repartidos en 3-5 días). En niños que reciben inmunodepresión primaria con ciclosporina, la conversión temprana a tacrolimus en caso de rechazo resistente a esteroides es el factor principal para evitar su progresión a rechazo crónico. El rechazo que aparece en niños con inmunodepresión primaria con tacrolimus habitualmente responde a esteroides; en los casos refractarios se emplea la asociación de micofenolato mofetil. En la experiencia de Pittsburgh, la inmunodepresión primaria con tacrolimus se asoció a la total ausencia de retrasplantes por rechazo agudo o crónico. En la experiencia europea reciente, el rechazo refractario ocurrió en el 1 y el 3% de los niños trasplantados que recibieron, respectivamente, tacrolimus o ciclosporina como inmunodepresión primaria39.
Afección biliar
La presencia de fugas de bilis, cuadros colestáticos obstructivos y colangitis son síntomas que derivan usualmente de una estenosis en la anastomosis quirúrgica y, más raramente, de estenosis o dehiscencias de la vía biliar de origen isquémico. Los injertos reducidos o segmentarios, más si son de donante vivo, tienen un riesgo aumentado respecto de los injertos enteros. La vía biliar de un lóbulo puede vascularizarse en algunas porciones por ramas arteriales procedentes del otro lóbulo. En otros casos, la justificación del problema es haber realizado la anastomosis biliar en la superficie de corte, a veces con varios conductos separados.
La frecuencia de afección biliar es de un 11% en el post- operatorio inmediato, casi restringida a los injertos reducidos. En ese período los problemas suelen requerir una reintervención. En el seguimiento a medio y largo plazo un 10% adicional de niños presenta estenosis biliar de forma que en los primeros 5 años la probabilidad acumulada de esta afección alcanza al 20% de los pacientes. La estenosis tardía afecta por igual a injertos enteros o reducidos28. Las manifestaciones clínicas ocurren por obstrucción e infección, ambas precipitadas por litiasis. El diagnóstico temprano es difícil, ya que pueden permanecer largo tiempo ocasionando una disfunción leve del injerto, sin evidente dilatación biliar en la ecografía ni cambios histológicos sugestivos de obstrucción biliar o colangitis. La colangiorresonancia magnética es mucho más eficaz para detectar este problema. La terapéutica por vía percutánea con dilatación de la estenosis y el empuje o triturado de los cálculos con dispositivos introducidos a través del catéter biliar hacen que sólo de manera excepcional sea necesaria la cirugía.
Disfunción idiopática del injerto
Las alteraciones en la función del injerto de causa no identificada se detectan con una prevalencia constante a lo largo de los años de seguimiento postrasplante de un 10%. Habitualmente se expresan histológicamente como focos de citólisis y analíticamente con elevación de transaminasas con o sin elevación de la gammaglutamil transpetidasa. En casos muy aislados ha ocurrido después fracaso del injerto por rechazo ductopénico o citólisis centrolobulillar severa y fue necesario efectuar un retrasplante tardío. La mayoría de los niños tiene una evolución estable y asintomática. Esta disfunción puede representar una forma de daño inmunológico o rechazo, de características histológicas y clínicas distintas de las que suceden en el postoperatorio inicial. La mayoría de los casos se tratan empíricamente con aumento de inmunodepresión.
Síndrome linfoproliferativo postrasplante
El VEB induce una proliferación de linfocitos B que en un individuo inmunocompetente es frenada por la respuesta de linfocitos T. Una vez curada la infección hay persistencia del virus en los tejidos, de forma latente. En los sujetos trasplantados que reciben inhibidores de calcineurina la respuesta de linfocitos T está disminuida. La falta de inhibición en la proliferación de linfocitos B inducida por VEB es la causa del desarrollo de PTLD.
Los sujetos que tienen primoinfección postrasplante son los más proclives a esta complicación. El hecho de que más de la mitad de los receptores de trasplantes hepáticos sean de edad muy pequeña, y que el hígado implantado proceda de sujetos de más edad, hace que la frecuencia de una situación de riesgo (receptor seronegativo y donante seropositivo frente al VEB) sea elevada.
En nuestra experiencia, desde 1986 hasta 1998, con ciclosporina como inmunodepresión primaria (en micro- emulsión desde 1995) y el uso de tacrolimus para rechazo resistente a los corticoides (desde 1992), la probabilidad acumulada de PTLD fue del 3,4% a los 2 años, del 8,8% a los 5 años y del 11,7% a los 10 años. En la experiencia de otros centros, con inmunodepresión primaria con tacrolimus, se han reportado incidencias globales del 2,4% (Japón), el 7% (Palo Alto), el 10% (UCLA) o el 14,6% (Lovaina); en Pittsburgh la tasa anual fue del 1,8%. En la experiencia de los últimos 5 años en nuestro hospital, en que se ha utilizado 3 meses de prevención con antivirales y se ha monitorizado la aparición de viremia por VEB para tratar a los positivos, la probabilidad acumulada de PTLD fue del 5,7% a los 12 meses, del 7,3% al tercer año y del 10,8% al quinto año, sin diferencia significativa entre los niños que recibieron ciclosporina en microemulsión o tacrolimus. Parece un hecho que esta complicación aparece más en la experiencia reciente, a pesar de las medidas de prevención. Es explicable por diversos factores, que incluyen el trasplante cada vez más predominante de niños muy pequeños, la mayor concienciación y búsqueda de este problema ante signos incipientes o inespecíficos y, sin duda, la mayor potencia de la inmunodepresión.
El PTLD es un síndrome clínico muy multiforme. En algunos niños hay datos tempranos, como neutropenia o hipergammaglobulinemia (pre-PTLD), sin un foco donde pueda realizarse una biopsia y documentar el proceso. La localización de PTLD más frecuente es amigdalar o adenoidea (fig. 4), pero puede darse en ganglios o en cualquier órgano (hueso, iris, tubo digestivo, hígado, pulmón, sistema nervioso central). Puede tratarse en otros casos de una afectación sistémica, con fiebre, esplenomegalia, pancitopenia, nefromegalia, neumopatía, etc.
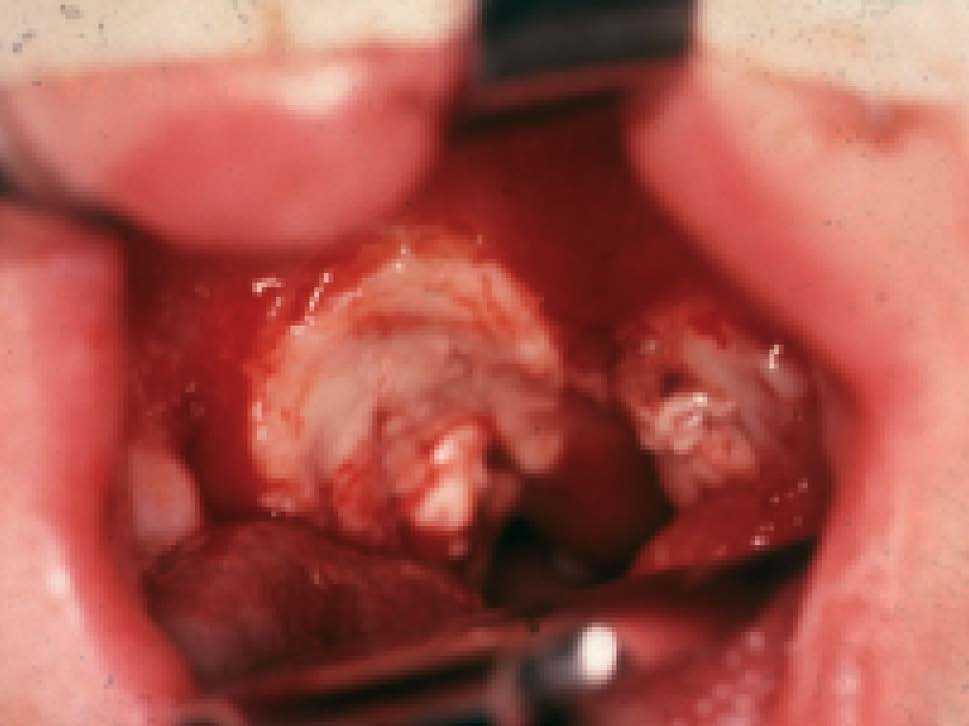
Fig. 4. Linfoma de alto grado que afecta a las amígdalas.
El diagnóstico se basa en los hallazgos histológicos que demuestran linfocitos de estirpe B (CD20+) con marcadores de infección por VEB (EBER positivo, o ADN-VEB positivo). La clasificación histológica del PTLD no es universal, por lo que resulta difícil comparar experiencias entre centros y los resultados del tratamiento. Los principales datos histológicos para evaluar un caso de PTLD son: a) la afectación de la estructura del órgano donde asienta (conservada o perdida); b) la morfología de la proliferación linfocitaria (monomorfa o polimorfa); c) el estudio del reordenamiento de inmunoglobulina H (policlonal o monoclonal), y d) el estudio de marcadores de malignidad (porcentaje de expresión de P53, alteraciones citogenéticas). Así, se distingue un espectro que abarca una «hiperplasia plasmacítica» como grado más leve a un «linfoma» como grado más severo50. No existe una correlación entre la clínica y la histología y si el paciente tiene varias localizaciones de PTLD pueden existir diferentes grados histológicos en ellas.
Los datos histológicos sirven para establecer una guía de terapéutica, según se considere que el proceso puede curar con el restablecimiento de la inmunidad de linfocitos T (mediante la disminución o retirada de los anticalcineurínicos) o es ya «maligno» en sentido estricto y susceptible de quimioterapia.
El tratamiento del PTLD no es uniforme entre centros. La reducción de la inmunosupresión es sin embargo el punto común de todas las actitudes51. Todos los procesos de PTLD consistentes en proliferaciones policlonales pueden revertir con la retirada de ciclosporina o tacrolimus, y mantener el esteroide. También los resultados avalan la conveniencia de resecar una lesión localizada. En la práctica ello se reserva para la afectación amigdalar exclusiva. El tratamiento antiviral con ganciclovir (preferible a aciclovir) es controvertido porque sólo actúa al impedir la fase lítica de la replicación del VEB y se conoce que la viremia que existe en los pacientes con PTLD procede en su mayor parte de los linfocitos B transformados por el VEB. El antiviral no afecta in vitro, y frena la proliferación de linfocitos B inducida por el VEB; sin embargo, se considera que mediante ganciclovir puede disminuir la infección nueva de linfocitos B. Se han empleado otros tratamientos sin que su eficacia pueda determinarse exactamente, como el interferón. Recientemente está disponible rituximab, un anticuerpo anti-CD20 que origina la destrucción mediada por complemento de los linfocitos B. La experiencia es incipiente y se han reportado remisiones junto con un elevado índice de pérdida de injerto por rechazo.
La aplicación de quimioterapia se reserva para pacientes con patrones histológicos compatibles con un linfoma, con proliferación monoclonal y expresión de alteraciones citogenéticas. Otros autores aplican quimioterapia en procesos no malignos en sentido estricto, pero sólo como un tratamiento inicial para conseguir la disminución de la linfoproliferación si hay riesgo de compromiso vital, por ejemplo por adenopatías o afección otorrinolaringológica que causan dificultad respiratoria.
Un 80% de los niños afectos de PTLD sobreviven. La retirada de la inmunodepresión puede ocasionar rechazo, en ocasiones antes de que el proceso linfoproliferativo haya remitido. La tardanza en aplicar tratamiento frente al rechazo, por ser prioritario el control de la PTLD, hace que en algunos pacientes sea necesario evolutivamente un retrasplante por rechazo crónico. No obstante, es más habitual que la retirada de la inmunodepresión pueda mantenerse meses o años, con remisión pronta del PTLD y sin que se produzca un rechazo. Un niño que ha presentado PTLD es un paciente complejo, que siempre podrá recaer una vez reintroducida la inmunodepresión; por ello sólo volverá a recibir anticalcineurínicos si se comprueba la aparición del rechazo.
El 20% de los pacientes afectos de PTLD fallece por complicaciones debidas a una grave afectación inicial, por no haberse diagnosticado adecuadamente o por que presentan linfomas de evolución refractaria a la quimioterapia.
RESULTADOS DEL TRASPLANTE
Aunque por lo que se ha expuesto se deduce la complejidad del curso posterior al trasplante hepático, de la posibilidad de morbilidad renal o del injerto a largo plazo, y del riesgo siempre presente de complicaciones graves, una vez pasado el postoperatorio inmediato la calidad de vida y el pronóstico de estos pacientes son generalmente muy buenos. Sobrevivir el primer año es un factor asociado a la supervivencia larga, con menos del 10% de mortalidad en los siguientes 10 años (fig. 5).
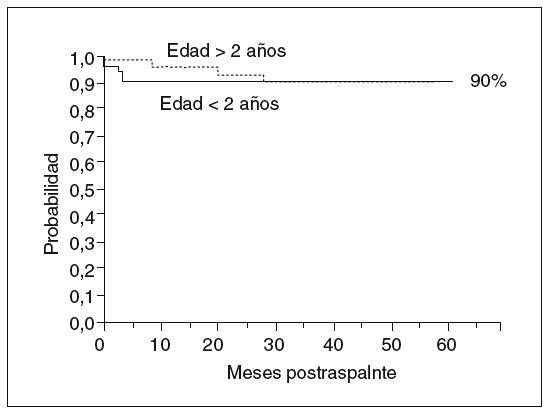
Fig. 5. Probabilidad de supervivencia en niños trasplantados. Experiencia reciente (1997-2001) Hospital Infantil La Paz. Respecto a épocas previas, resalta la buena supervivencia de los niños pequeños.

