








Introducción
El cáncer es una de las enfermedades más comunes y graves en el hombre. Los datos estadísticos muestran que un tercio de la población está afectada de alguna forma de cáncer, es causante del 20% de todas las muertes y, en países desarrollados, es causa de más del 10% del gasto sanitario. El diagnóstico y tratamiento tempranos, así como la identificación mediante técnicas de biología y genética moleculares de los individuos con una mayor probabilidad de desarrollarlo antes de que ello ocurra, son objetivos prioritarios de la investigación biomédica actual1.
El desarrollo del cáncer colorrectal (CCR) comporta una serie de cambios histopatológicos en la mucosa colorrectal bien caracterizados y conocidos, como la secuencia adenoma-carcinoma2. Esta progresión es el resultado de una serie de cambios genéticos secuenciales que consisten en la activación de oncogenes y la inactivación de genes supresores de tumor3,4, así como cambios epigenéticos5.
El CCR es una de las enfermedades más frecuentes en las poblaciones occidentales y la segunda causa de muerte por cáncer. En Europa se diagnostican cada año 215.000 casos nuevos, que ocasionan más de 110.000 muertes. En España, con una incidencia de 30-35 casos por 100.000 habitantes, representa el segundo tumor más frecuente tras el cáncer de pulmón y la segunda causa de muerte por cáncer6,7. El riesgo de desarrollar CCR es del 5% en la población general, aunque esta cifra aumenta exponencialmente con la edad8 y es mucho más elevado en individuos con una historia familiar de CCR.
En el CCR existe un gran componente hereditario o familiar, el cual podría ser mayor que en cualquier otra enfermedad humana adulta. Así, se ha estimado que el 20-30% de los casos de CCR afectan a individuos predispuestos genéticamente9. Sin embargo, en la actualidad tan sólo un pequeño porcentaje puede atribuirse a un síndrome hereditario específico10.
Los síndromes de CCR hereditario conocidos se subdividen en los asociados o no a poliposis11,12. Los síndromes polipósicos se subdividen a su vez, en función del tipo histológico, en adenomatosos y hamartomatosos. Los síndromes de CCR asociados a poliposis representan el 0,5% del total de casos y los adenomatosos son mucho más frecuentes que los hamartomatosos. Por el contrario, el CCR no asociado a poliposis representa un 1-5% del total. Finalmente, los casos familiares que no corresponden a los síndromes antes descritos constituyen un 20-25% de la totalidad de CCR, aunque esta fracción podría ser mucho mayor según se deduce del estudio de genealogías10,13.
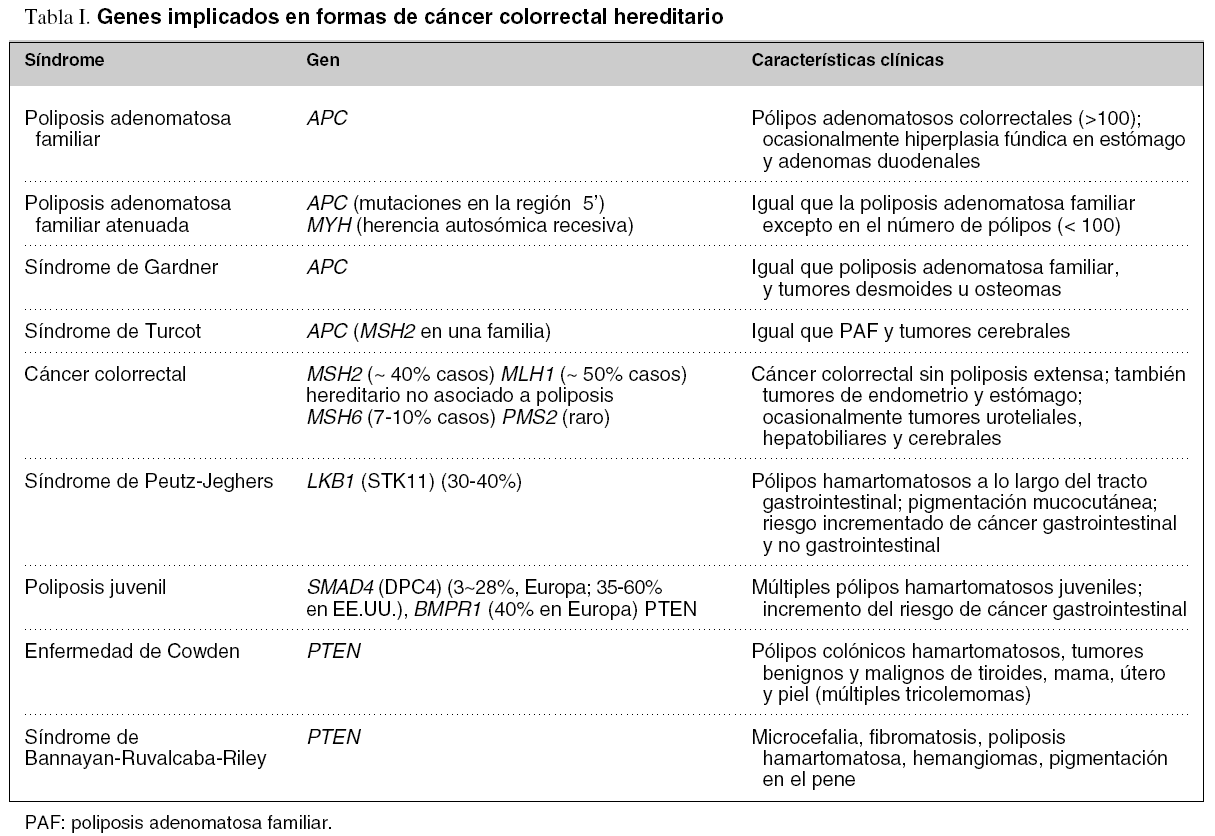
En los últimos años se han identificado muchos de los genes responsables de las distintas formas hereditarias de CCR (tabla I), lo que ha abierto la posibilidad del diagnóstico molecular para este subgrupo de pacientes6,14,15. Dada su mayor prevalencia, la presente revisión se centrará en la poliposis adenomatosa familiar y el CCR hereditario no asociado a poliposis.
Poliposis adenomatosa familiar
La poliposis adenomatosa familiar (PAF) es un síndrome hereditario de predisposición genética al CCR en el cual se desarrollan múltiples adenomas (habitualmente más de 100) distribuidos a lo largo del colon y/o recto. La enfermedad acostumbra presentarse alrededor de los 16 años de edad (rango: 7-36 años) y los pólipos están presentes en más del 95% de los pacientes a los 35 años. Además, la aparición de CCR es inevitable si no se realiza una colectomía profiláctica; la edad media de presentación en los individuos no tratados es de 39 años (rango: 34-43 años). Alrededor del 60% de los pacientes presentan lesiones gastroduodenales asociadas, como poliposis glandular fúndica, pólipos hiperplásicos o adenomas, y es muy frecuente la existencia de lesiones hiperpigmentadas de la retina16. Se estima que esta enfermedad afecta a uno de cada 15.000 individuos y presenta una penetración prácticamente del 100%.
La PAF se hereda de forma autosómica dominante y está causada por mutaciones en el gen supresor APC17. El gen APC es de un tamaño considerable, se compone de 15 exones que codifican para una proteína de 2.843 aminoácidos implicada en la adhesión celular, la transducción de señal y la activación transcripcional.
La PAF es un ejemplo de cáncer hereditario en el cual las variaciones alélicas de un mismo gen dan lugar a una diversidad en el fenotipo (tabla I). Así, se ha descrito una variante denominada poliposis adenomatosa familiar atenuada, que se caracteriza por un inicio más tardío y un menor número de pólipos localizados de manera preferente en el colon derecho, y cuya alteración molecular consistiría en la presencia de mutaciones en el extremo 5' del gen APC. Por otra parte, la identificación de APC ha permitido confirmar que el síndrome de Gardner forma parte del espectro clínico de la PAF. En este sentido, se reserva esta denominación para los individuos que presentan lesiones extraintestinales asociadas a la afectación colorrectal. Dado que los pólipos son de tipo adenomatoso, el riesgo de malignización es idéntico al descrito en la PAF. Las lesiones extraintestinales más frecuentes son osteomas en maxilares, cráneo y huesos largos, tumores desmoides, fibromatosis mesentérica y quistes subdentarios. Por último, el síndrome de Turcot es una enfermedad hereditaria autosómica recesiva inicialmente descrita como la asociación de PAF y tumores del sistema nervioso central. Sin embargo, recientemente se ha descrito la presencia de mutaciones germinales tanto en el gen APC como en un gen responsable de la reparación del ADN (MSH2). Por ello, los tumores del sistema nervioso central deberían incluirse entre las lesiones asociadas tanto a la PAF como al CCR hereditario no asociado a poliposis.
En la actualidad, gracias a que se conoce el defecto molecular en el gen APC, existe la posibilidad de realizar un análisis genético para la confirmación diagnóstica o la identificación de portadores asintomáticos en familiares con riesgo en el caso de que se conozca la mutación causante en una determinada familia con PAF. Además, existe cierta correlación genotipo-fenotipo en el sentido de que la localización de la mutación en APC puede determinar el espectro clínico de la enfermedad18.
Las técnicas empleadas para el diagnóstico genético de PAF son variadas y incluyen la técnica PTT (protein truncation test, prueba de la proteína truncada), que permite detectar la mayoría de las mutaciones identificadas en este gen, las cuales comportan una terminación prematura de la proteína. Otras técnicas empleadas para el cribado mutacional del gen APC son SSCP (single strand conformation polymor-phism), DGGE (denaturing gradient gel electrophoresis), DHPLC (denaturing high-performance liquid chromatography) y la secuenciación. Además, también pueden aplicarse técnicas como el análisis Southern blot o la técnica MLPA (multiplex ligation-dependent probe amplification), que permiten detectar deleciones o duplicaciones en este gen. Por último, existe la posibilidad de realizar un diagnóstico genético indirecto mediante análisis de ligamiento en familias con PAF con más de un miembro afectado mediante marcadores altamente polimórficos del gen APC (microsatélites) y así establecer el haplotipo ligado a la enfermedad. Este enfoque, sin embargo, no permite identificar la mutación causante. En la tabla II se muestra el rendimiento de las diferentes técnicas para la detección de mutaciones en el gen APC, mientras que en la figura 1 se esquematiza la estrategia de diagnóstico molecular en la PAF que se emplea en nuestro centro.

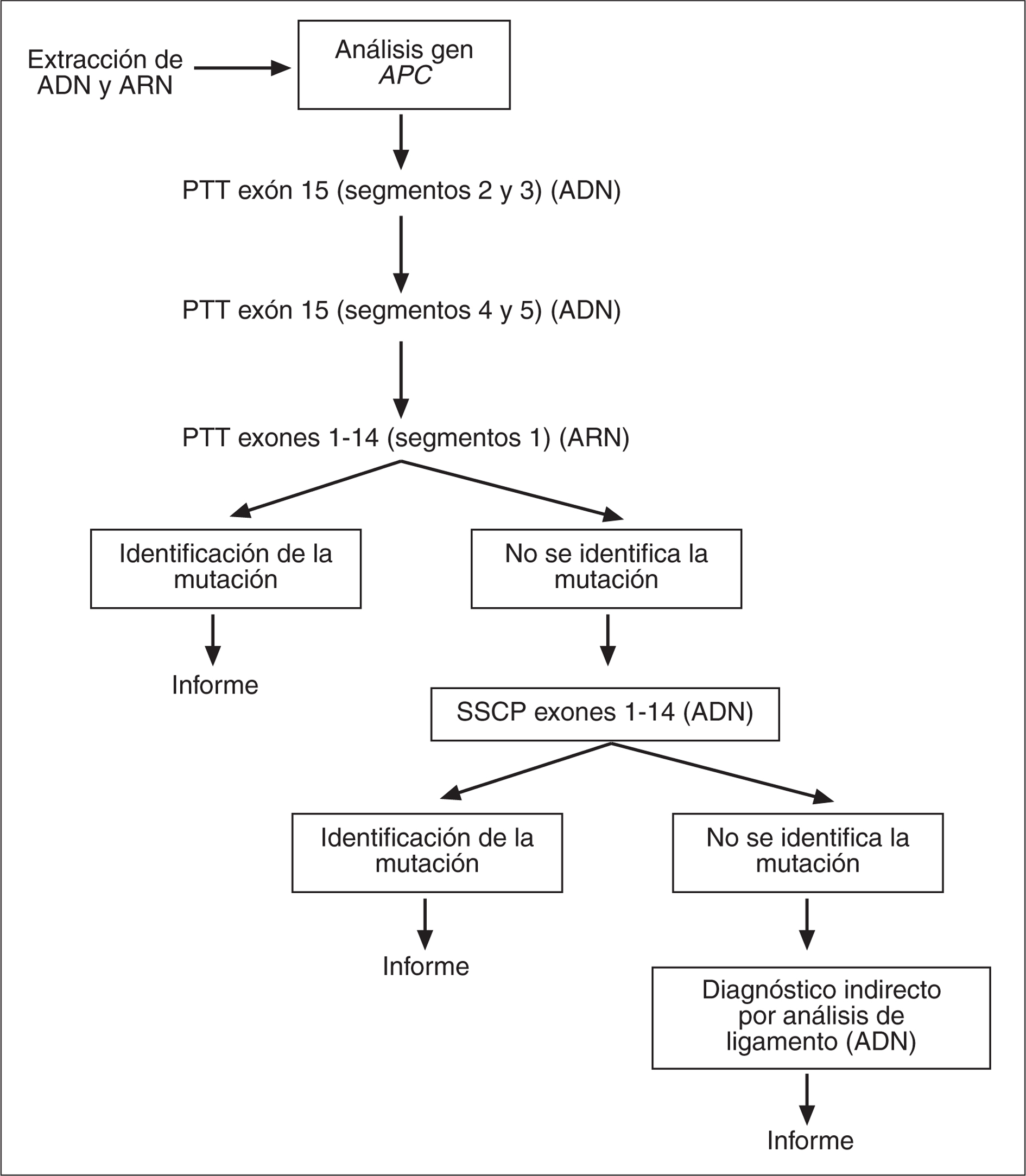
Fig. 1. Estrategia de diagnóstico molecular en la poliposis adenomatosa familiar. PTT: protein truncation test; SSCP: single strand conformation poly- morphism.
Recientemente, se han descrito mutaciones germinales en el gen MYH en pacientes con múltiples adenomas colorrectales o PAF en los que no se habían identificado mutaciones en el gen APC19. El gen MYH está implicado en la reparación del ADN por escisión de bases y actúa para paliar el daño oxidativo al ADN. Este gen parece actuar de forma autosómica recesiva con mutaciones bialélicas necesarias para la expresión del fenotipo, a diferencia de APC, que actúa de forma autosómica dominante, y está implicado en una proporción significativa de pacientes con PAF atenuada.
Una vez identificada la mutación patogénica en APC o MYH en una determinada familia, se ha de ofrecer la posibilidad de diagnóstico de confirmación y/o presintomático al resto de los miembros de la familia siguiendo las pautas correctas del consejo genético (véase más adelante).
Cáncer colorrectal hereditario no asociado a poliposis
El CCR hereditario no asociado a poliposis (CCHNP) es una enfermedad hereditaria autosómica dominante y corresponde a la predisposición genética más común a desarrollar CCR u otras neoplasias como cáncer de endometrio, ovario, estómago, intestino delgado, tracto hepatobiliar, tracto urinario superior, cerebro y piel20. El CCHNP constituye entre el 1 y el 3% de los casos de cáncer CCR dependiendo de la población estudiada. En la población española se estima que representa el 2,5%10.
El CCHNP está asociado a mutaciones germinales en genes implicados en la vía de reparación del emparejamiento incorrecto del ADN (mismatch repair, MMR), en especial MLH1, MSH2, MSH6 y PMS2. Las mutaciones en MLH1 y MSH2 son las mayoritarias y suponen alrededor del 90% de las mutaciones identificadas en familias con CCHNP, mientras que las mutaciones en MSH6 suponen alrededor del 7-10% y las mutaciones en PMS2 representan menos del 5%. La penetración del CCR asociado a mutaciones en estos genes es del 80% aproximadamente. También se han publicado mutaciones en los genes MSH3, EXO1 y TGFßR2 en algunas familias con CCHNP, aunque su relevancia clínica no está bien establecida21. La base de datos de mutaciones de la International Society for Gastrointestinal Hereditary Tumours (InSiGHT) (http://www.insight-group.org) contiene, en su última actualización, 448 mutaciones identificadas en 748 familias de todo el mundo.
Las causas más habituales de consulta genética por CCHNP son una historia familiar de CCR o neoplasias extracolónicas anteriormente citadas, una historia personal de tumores sincrónicos o metacrónicos y/o la aparición temprana de CCR en ausencia de poliposis.
Debido a la elevada incidencia de CCR, es posible observar una agregación de tumores presumiblemente esporádicos en el seno de una determinada familia sin que eso signifique que nos encontramos ante un caso de CCHNP. De esa manera, debido a que no es factible efectuar el análisis genético en todos los enfermos afectados de CCR, se recomienda hacer una selección clínica minuciosa de las familias con CCHNP previa al análisis molecular basándose en criterios clínicos establecidos. Dado que los criterios de Amsterdam originales22 o revisados23 son muy específicos pero poco sensibles, se ha propuesto utilizar los criterios de Bethesda24, recientemente revisados25.
Para llevar a cabo el análisis molecular se sigue un algoritmo como el esquematizado en la figura 2. Una vez identificado un caso índice de CCHNP con los criterios clínicos anteriormente mencionados, se procede a efectuar el análisis molecular14,26,27. Si se dispone de muestra tumoral, se pueden utilizar las técnicas de inestabilidad de microsatélites y/o la inmunohistoquímica para las proteínas reparadoras28. A nivel molecular, la inestabilidad de microsatélites constituye un marcador fenotípico del CCHNP aunque no exclusivo, ya que también se puede encontrar en el 7-15% de los tumores CCR esporádicos debido a hipermetilación del promotor de MLH1 o mutaciones somáticas en algún gen reparador.
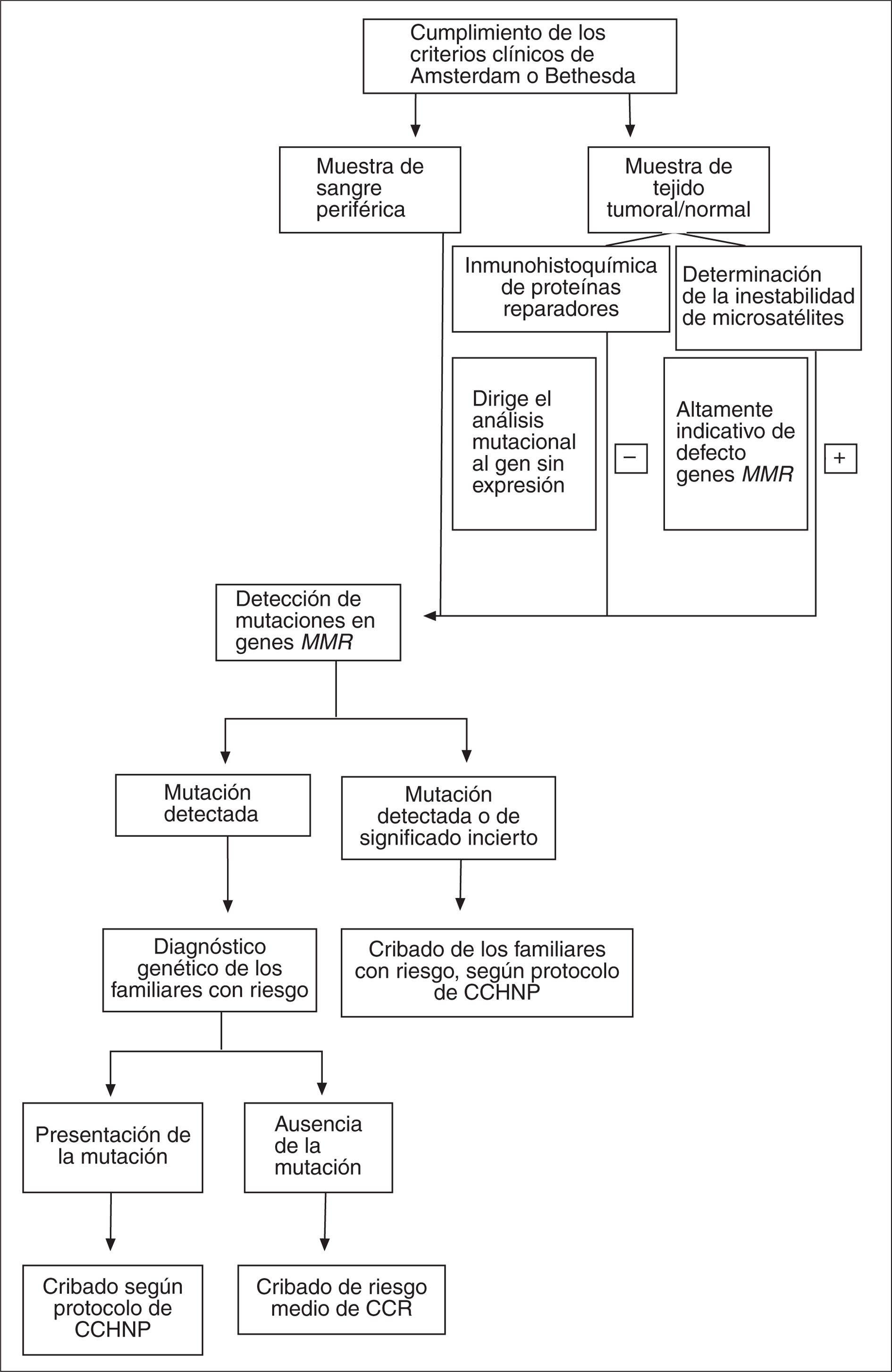
Fig. 2. Estrategia de cribado y diagnóstico molecular del cáncer colorrectal hereditario no asociado a poliposis. MMR: reparación del emparejamiento incorrecto del ADN (mismatch repair); CCHNP: cáncer colorrec- tal hereditario no asociado a poliposis; CCR: cáncer colorrectal.
La inmunohistoquímica para las proteínas reparadoras MLH1, MSH2, MSH6 y PSM2 en tejido tumoral detecta la presencia o ausencia de la proteína reparadora analizada y, en caso de ausencia, permite dirigir el análisis molecular posterior. Sin embargo, algunas mutaciones en genes reparadores no dan lugar a la ausencia de la proteína correspondiente. Este análisis puede complementar al análisis de inestabilidad de microsatélites o, incluso, ser una alternativa a éste en los centros que no disponen de laboratorio para la realización de técnicas moleculares.
En cuanto al análisis mutacional en los genes reparadores, se realiza mediante la detección de mutaciones germinales en ADN genómico. Actualmente se recomienda realizar la detección de reordenamientos genómicos en los genes reparadores antes del cribado de mutaciones concretas, debido a la relativa elevada frecuencia de estas alteraciones en familias con CCHNP y a su simplicidad técnica gracias a la disponibilidad de ensayos rápidos, como la técnica MLPA (fig. 3). En caso de detectar un reordenamiento genómico se obvia el cribado posterior29.
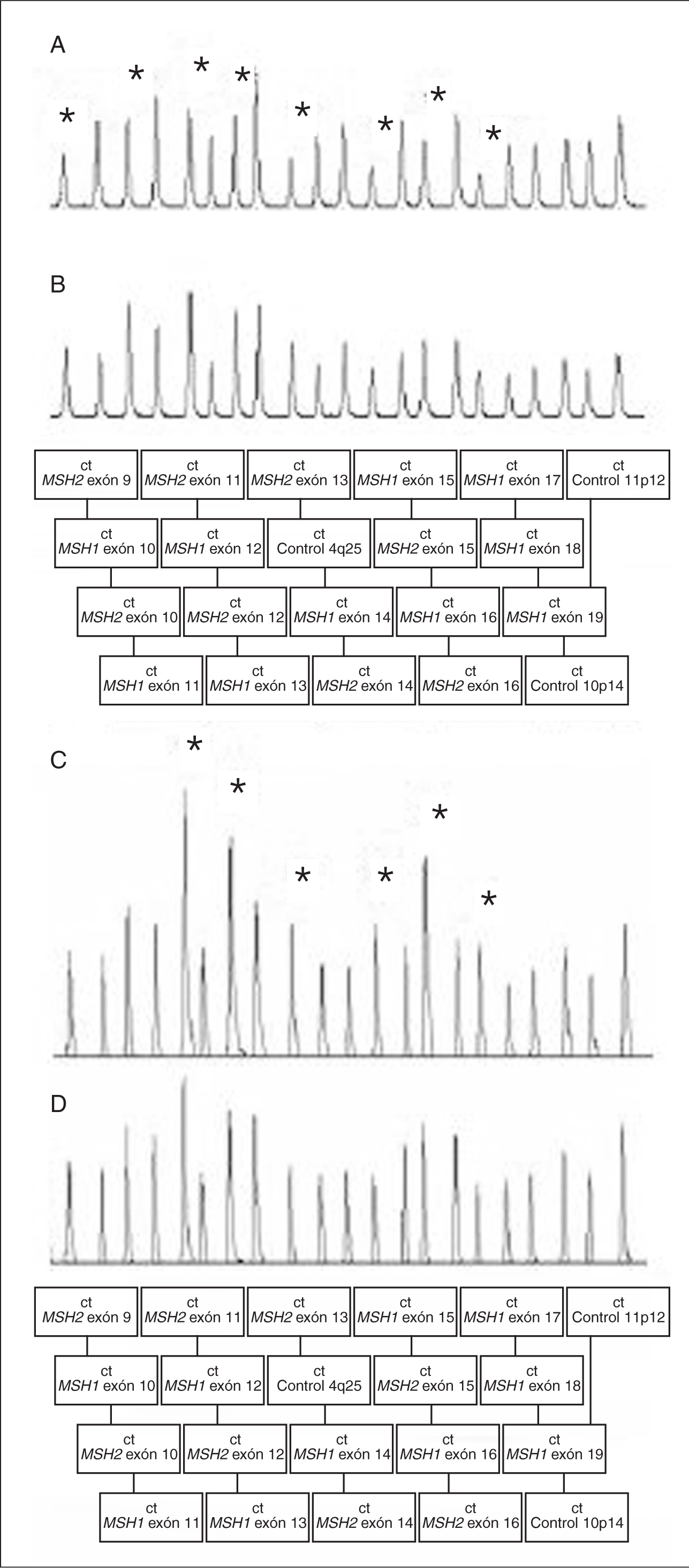
Fig. 3. MLPA (multiplex ligation-dependent probe amplification). Cromatogramas en muestras con deleción de los exones 9 al 16 en MSH2 (A) o duplicación de los exones 11 al 16 en MSH2 (C), y en muestra de controles (B y D). Los asteriscos indican los fragmentos delecionados o duplicados en las muestras A y C.
El cribado de mutaciones concretas en los genes reparadores implicados se puede efectuar mediante diversas técnicas (secuenciación completa, PTT, SSCP, DHPLC, DGGE u otras), dependiendo de las preferencias y disponibilidad del laboratorio. Este cribado se suele realizar exón por exón o en varios fragmentos de cada gen reparador y es, por tanto, una técnica costosa y laboriosa. Si no se ha utilizado la secuenciación completa como estrategia de cribado, una vez detectado un patrón anómalo, se debe caracterizar la mutación causante mediante esta técnica en el ADN original. En la tabla II se muestra el rendimiento de las diferentes técnicas para el diagnóstico molecular del CCHNP.
Una vez identificada la mutación patogénica en uno de los genes reparadores en una determinada familia, se ha de ofrecer la posibilidad de diagnóstico de confirmación y/o presintomático al resto de los miembros siguiendo las pautas correctas del consejo genético (véase más adelante).
Consejo genético en el cáncer colorrectal hereditario
Los nuevos conocimientos sobre las causas genéticas del CCR hereditario no sólo tienen importancia para el diagnóstico y el manejo de los pacientes, sino también para la estimación del riesgo de padecer la enfermedad y el consejo genético de las familias.
Es muy aconsejable una evaluación psicológica de los individuos en los cuales se va a proceder al análisis genético con el fin de valorar el impacto que pueden tener sus resultados. Asimismo, la estimación de riesgo mediante pruebas predictivas debe incluir siempre sesiones educativas o de consejo previas llevadas a cabo por personal especializado con el objetivo de informar ampliamente sobre los aspectos hereditarios de la enfermedad, las opciones de diagnóstico genético y sus posibles implicaciones. Del mismo modo, cuando se informe del resultado de la prueba genética debe hacerse con las máximas garantías de claridad y comprensión por parte del paciente, así como realizar una posterior sesión informativa en el caso de una prueba positiva con el fin de resolver cualquier duda referente a la enfermedad.

