








De todas las neoplasias digestivas hereditarias, las más importantes por su frecuencia son las que afectan al colon y recto, principalmente el síndrome de Lynch y la poliposis adenomatosa familiar. Sin embargo existen otros grupos de neoplasias digestivas extracolónicas muy poco estudiadas y conocidas, que constituyen un pequeño porcentaje de los cánceres hereditarios digestivos. A pesar de ser poco frecuentes, estas neoplasias merecen importancia debido a la gran morbimortalidad que conllevan, destacando principalmente el adenocarcinoma gástrico y pancreático. Este artículo tiene como objetivo hacer una revisión de los datos conocidos hasta la fecha de los síndromes hereditarios y familiares asociados a estas dos neoplasias, de cara a un mayor conocimiento y entendimiento de estas patologías, con la intención de mejorar la sospecha diagnóstica y así poner en marcha las estrategias diagnósticas, de cribado, de vigilancia y terapéuticas adecuadas.
The most common hereditary gastrointestinal cancers are colorectal, mainly hereditary nonpolyposis colorectal cancer (Lynch syndrome) and familial adenomatous polyposis. Other extracolonic neoplasms, including the gastric and pancreatic adenocarcinomas, are less well known and studied because they account for a relatively small percentage of hereditary gastrointestinal cancers. Nonetheless, they merit special attention because of the high associated morbidity and mortality rates. We review the hereditary and familial syndromes associated with gastric and pancreatic cancers with a view to improving knowledge and understanding of these diseases, in order to heighten diagnostic suspicion and thus implement appropriate diagnostic strategies, screening, surveillance and treatment.
El cáncer gástrico (CG) es el quinto cáncer más frecuente en el mundo y una de las principales causas de muerte oncológica. En España, la incidencia es de 7,8 casos por 100.000 habitantes, siendo dos veces más frecuente en hombres que en mujeres1. La edad media al diagnóstico es de 60 años. Tan solo un 7% se presentan antes de los 50 años y un 2% antes de los 40 años2.
La etiología del CG es multifactorial, entre los agentes ambientales principales se encuentran Helicobacter pylori (H. pylori), la dieta y el tabaco. Histológicamente, se clasifica principalmente en adenocarcinoma tipo intestinal y difuso. El CG intestinal se asocia más estrechamente a factores ambientales y a edad avanzada, mientras que el CG difuso ocurre en personas más jóvenes y se caracteriza por un infiltrado multifocal con células en anillo de sello2.
Si bien la mayoría de los CG son esporádicos, se observa una agregación familiar en aproximadamente el 10% de los casos2,3. El CG hereditario, es decir, en contexto de una mutación germinal asociada, representa un 1-5% de todos los CG4. Entre las características familiares que sugieren una predisposición hereditaria se incluyen el presentar varios familiares afectos, un patrón de herencia autosómico dominante, afectación a edades tempranas y la asociación a otras neoplasias extragástricas.
En base a estos parámetros, existen tres situaciones clínicas en las que se puede encontrar predisposición familiar al CG:
- 1)
Síndromes hereditarios con mayor riesgo principalmente de CG: este escenario incluye a dos entidades:
1.1 Cáncer gástrico difuso hereditario (CGDH) y, 1.2 adenocarcinoma gástrico asociado a poliposis proximal en estómago (AGPPE).
- 2)
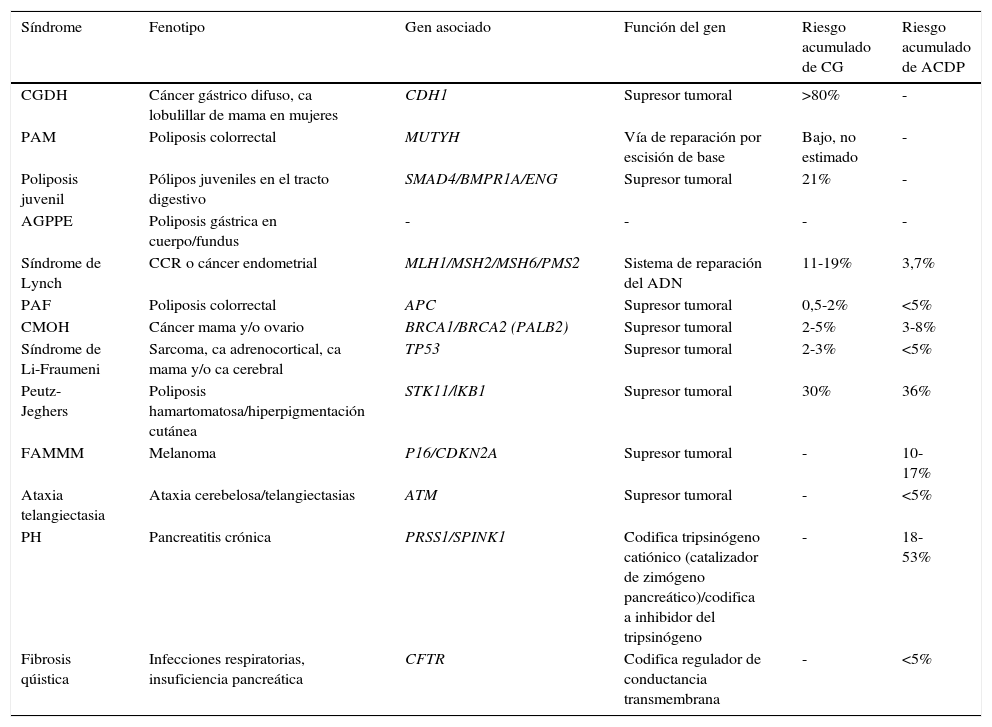
Síndromes hereditarios con mayor riesgo de CG y otros tumores: en contexto de otros síndromes de cáncer hereditario que asocian un mayor riesgo de CG y de otras neoplasias, con una mutación germinal asociada (tabla 1).
Tabla 1.Síndromes de cánceres hereditarios asociados a adenocarcinoma gástrico y adenocarcinoma ductal de páncreas
Síndrome Fenotipo Gen asociado Función del gen Riesgo acumulado de CG Riesgo acumulado de ACDP CGDH Cáncer gástrico difuso, ca lobulillar de mama en mujeres CDH1 Supresor tumoral >80% - PAM Poliposis colorrectal MUTYH Vía de reparación por escisión de base Bajo, no estimado - Poliposis juvenil Pólipos juveniles en el tracto digestivo SMAD4/BMPR1A/ENG Supresor tumoral 21% - AGPPE Poliposis gástrica en cuerpo/fundus - - - - Síndrome de Lynch CCR o cáncer endometrial MLH1/MSH2/MSH6/PMS2 Sistema de reparación del ADN 11-19% 3,7% PAF Poliposis colorrectal APC Supresor tumoral 0,5-2% <5% CMOH Cáncer mama y/o ovario BRCA1/BRCA2 (PALB2) Supresor tumoral 2-5% 3-8% Síndrome de Li-Fraumeni Sarcoma, ca adrenocortical, ca mama y/o ca cerebral TP53 Supresor tumoral 2-3% <5% Peutz-Jeghers Poliposis hamartomatosa/hiperpigmentación cutánea STK11/lKB1 Supresor tumoral 30% 36% FAMMM Melanoma P16/CDKN2A Supresor tumoral - 10-17% Ataxia telangiectasia Ataxia cerebelosa/telangiectasias ATM Supresor tumoral - <5% PH Pancreatitis crónica PRSS1/SPINK1 Codifica tripsinógeno catiónico (catalizador de zimógeno pancreático)/codifica a inhibidor del tripsinógeno - 18-53% Fibrosis qúistica Infecciones respiratorias, insuficiencia pancreática CFTR Codifica regulador de conductancia transmembrana - <5% AGPPE: adenocarcinoma gástrico asociado a poliposis proximal en estómago; Ca: cáncer; CCR: cáncer colorrectal; CGDH: cáncer gástrico difuso hereditario; CMOH: síndrome de cáncer de mama-ovario hereditario; FAMMM: melanoma maligno múltiple atípico familiar; PAF: poliposis adenomatosa familiar; PAM: poliposis adenomatosa asociada a MUTYH; PH: pancreatitis hereditaria.
- 3)
Cáncer gástrico familiar: situación en la que se observa una agregación familiar de CG de histología intestinal sin una causa genética identificada.
Esta entidad representa entre el 1-3% de todos los CG. Inicialmente descrita en 1998 con la identificación de una mutación germinal en el gen CDH1 en familias de origen maorí con múltiples casos de CG difuso5. Desde entonces, se han identificado mutaciones en CDH1 hasta en el 50% de las familias con criterios clínicos de CGDH. Un estudio6 publicado recientemente analizó la serie más larga de portadores de mutaciones en CDH1 reportada hasta ahora, objetivando un riesgo acumulado de CG del 70% a los 80 años en hombres portadores de mutación en CDH1, y de 56% en mujeres, así como un riesgo acumulado de cáncer de mama en mujeres del 42% a los 80 años. Así mismo, también se ha establecido un potencial riesgo incrementado de cáncer colorrectal y prostático7,8.
Genética. Patrón de herencia autosómico dominante. El CGDH está asociado a mutaciones germinales en el gen CDH1 en el 30-50% de los casos. El gen CDH1 se localiza en el cromosoma 16q22.1 y codifica para la proteína E-cadherina. El gen CDH1 actúa como un gen supresor de tumores y su mutación conduce a la pérdida de adhesión celular, favoreciendo la proliferación, invasión y metástasis. En los individuos con una mutación germinal en CDH1 se produce la inactivación somática del alelo sano de CDH1 favoreciendo la aparición de tumores en las localizaciones descritas.
Se han identificado más de 100 mutaciones germinales de CDH1. La mayoría de las mutaciones son truncadas (principalmente de corrimiento del marco de lectura [frameshift], en los sitios de corte y empalme [splice-site] y mutaciones sin sentido [nonsense]), seguidas de las mutaciones de cambio de aminoácido (missense)9,10. Adicionalmente, también se han identificado mutaciones en el gen alfa-E-catenina (CTNNA1) como causa de CGDH11.
La penetrancia es alta, aunque variable, se estima de 63-83% en mujeres y de 40-67% en hombres.
Anatomía patológica. Estos tumores se caracterizan por una histología difusa con células en anillo de sello (fig. 1); los focos tumorales suelen ser múltiples y propagarse a través de toda la mucosa gástrica con una mayor concentración en el antro y en la zona de transición entre el antro y cuerpo.
.")
Criterios diagnósticos. De acuerdo al International Gastric Cancer Linkage Consortium de 2004 actualizado en 2015 los criterios que definen al CGDH y por tanto las situaciones en las que está indicado realizar análisis genético germinal (estudio de mutaciones en el gen CDH1) son los siguientes12:
- •
Familia con>2 casos de cáncer gástrico difuso (CGD), al menos uno de los casos confirmado, con familiares de primer grado (FPG) o familiares de segundo grado (FSG), con independencia de la edad de diagnóstico, o
- •
Individuos con CGD diagnosticado antes de los 40 años sin historia familiar, o
- •
Antecedente personal o familiar de CGD y cáncer de mama lobulillar, alguno de ellos diagnosticado antes de los 50 años.
Para evaluar de forma correcta que una familia cumpla los criterios de CGDH es esencial el informe de anatomía patológica preferiblemente valorado por un patólogo experto en cáncer gástrico. Los casos confirmados de cáncer gástrico de tipo intestinal no forman parte del CGDH, por ello, en estas familias no está indicado realizar análisis de mutación de CDH1.
Por otro lado, en las siguientes familias se debe considerar la realización de análisis genético12:
- •
Cáncer de mama lobulillar bilateral o historia familiar de>2 casos de cáncer de mama lobulillar<50 años.
- •
Historia personal o familiar de labio/paladar leporino en un paciente con CGD.
- •
Células en anillo de sello in situ y/o propagación de células en anillo de sello.
Prevención y cribado CG:
*Gastrectomía profiláctica. La mayoría de los pacientes asintomáticos portadores de una mutación germinal en CDH1 no tienen lesiones macroscópicas en las exploraciones endoscópicas, sin embargo, en las piezas quirúrgicas se objetivan focos intramucosos de CGD, habitualmente múltiples13. Por tanto, se recomienda realizar gastrectomía total profiláctica en portadores de una mutación patogénica, mayores de 20 años, recomendándose la cirugía 5 años antes de la edad del CG del familiar afecto más joven (la edad media al diagnóstico de CGD es de 38-40 años)7,14.
*Cribado endoscópico. Se reserva para pacientes que no optan por la gastrectomía profiláctica, pacientes con variante de significado incierto, y pacientes en los que no se ha podido identificar la mutación germinal. Se establece de acuerdo al protocolo de Cambridge14:
- •
Endoscopio de alta definición, inspección cuidadosa durante 30 min, maniobras de tensión/distensión gástrica, uso de mucolíticos, biopsia de todas las anomalías mucosas, biopsias seriadas: se recomiendan 30 biopsias aleatorias, 6 de cada región (antro, incisura, cuerpo, fundus y cardias), detección y erradicación de H. pylori.
El cribado con endoscopia se recomienda realizarlo cada 6-12 meses, iniciándose a los 20 años o 5 años antes de la edad del familiar con CG más joven en pacientes con mutación conocida y que rechazan la gastrectomía, y a partir de los 40 años o 5 años antes de la edad del familiar afecto más joven en los casos sin mutación patogénica identificada12,14 (fig. 2).

Otras neoplasias. Se recomienda la realización de RM mamaria (que podría ser combinada con mamografía) anual a partir de los 30 años de edad en mujeres portadoras de mutación germinal en CDH112. Además es esencial el examen mamario clínico anual. En familias con mutación conocida y casos de cáncer colorrectal se aconseja la realización de colonoscopia cada 3-5 años a partir de los 40 años o 10 años antes del familiar afecto más joven12.
Adenocarcinoma gástrico y poliposis proximal en estómagoEs un síndrome recientemente descrito de poliposis gástrica proximal con un riesgo elevado de CG tipo intestinal. La primera familia identificada fue en Australia y posteriormente se han identificado en otras familias de Estados Unidos y Canadá. Los pacientes presentan típicamente poliposis de glándulas fúndicas con áreas de displasia o adenocarcinoma gástrico tipo intestinal restringido al estómago proximal y sin evidencia de poliposis duodenal ni colónica. La edad de presentación del CG es variable (se han identificado casos desde los 33 hasta los 75 años), con un efecto de anticipación4,15.
Genética. Herencia autosómica dominante. Penetrancia incompleta. No se ha identificado el defecto genético causal.
Anatomía patológica. Se caracteriza por una poliposis gástrica florida (habitualmente>100 pólipos) menores de 10mm de diámetro, que tapizan el cuerpo y el fundus gástrico. Habitualmente el esófago, antro gástrico, píloro y duodeno están respetados. La histología es compatible con poliposis de glándulas fúndicas incluyendo áreas de displasia y ocasionalmente pólipos hiperplásicos y adenomatosos. La histología del cáncer es de tipo intestinal4,15,16.
Criterios diagnósticos. El diagnóstico se basa en la presencia de los siguientes criterios15:
- •
Poliposis gástrica de cuerpo o fundus sin evidencia de poliposis colorrectal o duodenal y >100 pólipos tapizando el estómago proximal en el caso índice o>30 pólipos en un FPG de un paciente con AGPPE y predominio de pólipos fúndicos, algunos con displasia y un familiar con pólipos fúndicos displásicos o CG y
- •
Patrón autosómico dominante.
Se debe distinguir de otros síndromes hereditarios como la poliposis adenomatosa familiar (PAF) clásica o atenuada, la poliposis asociada a MUYTH (PAM) y el síndrome de Peutz-Jeghers (SPJ). El uso de inhibidores de la bomba de protones también forma parte del diagnóstico diferencial, por lo cual para hacer el diagnóstico de AGPPE se debe excluir el uso de los mismos y repetir la endoscopia tras suspender este tratamiento para descartar su asociación.
Prevención y cribado de CG. Debido a las pocas familias identificadas hasta la fecha, el manejo no está bien establecido. Se recomienda individualizar riesgos/beneficios de la gastrectomía profiláctica y cribado endoscópico, teniendo en consideración las limitaciones de la vigilancia endoscópica en cada caso y el riesgo de CG de cada familia. Se recomienda la realización de gastroscopia y colonoscopia en los FPG de los individuos afectos4.
Síndromes hereditarios con mayor riesgo de cáncer gástrico y otros tumoresEl CG puede presentarse en contexto de otros síndromes de predisposición a cáncer como síndrome de Lynch (SL), Li-Fraumeni, SPJ, PAF, PAM, poliposis juvenil, y síndrome de cáncer de mama y ovario hereditario (CMOH)17 (tabla 1).
Síndrome de Lynch (cáncer colorrectal hereditario no polipósico)El SL se asocia principalmente a un incremento en el riesgo de cáncer colorrectal, sin embargo también conlleva riesgo de otras neoplasias. Dentro de las neoplasias del espectro Lynch destaca el cáncer de endometrio pero también se encuentran las de ovario, estómago, vía biliar, intestino delgado, páncreas, uréter y pelvis renal, así como el cáncer de piel (tumores sebáceos, en la variante conocida como síndrome de Muir-Torre) y los tumores del sistema nervioso central (glioblastomas y astrocitomas, en la variante conocida como síndrome de Turcot). El riesgo de CG en este contexto se estima entre el 6-13%, con una edad media al diagnóstico de 56 años18,19.
Genética. Es un síndrome con herencia autosómica dominante, causado por mutaciones a nivel germinal en alguno de los genes del sistema de reparación del ADN (MLH1, MSH2, MSH6 y PMS2). MLH1 está localizado en el cromosoma 3p21, MSH2 en el cromosoma 2p16, MSH6 en el cromosoma 2p16, y PMS2 en el cromosoma 7p22. El papel de los genes reparadores del ADN es mantener la integridad del genoma corrigiendo errores de sustitución de bases y pequeñas inserciones-deleciones que se generan durante la replicación del ADN, y que pueden afectar la regulación del crecimiento celular y promover la formación de neoplasias20.
Anatomía patológica. El CG en contexto del SL, es de tipo intestinal en el 90% de los casos y solo un pequeño porcentaje presenta una histología difusa. Las mutaciones germinales en los genes reparadores del ADN tienen dos consecuencias somáticas detectables en el seno del tumor que desarrollan estos pacientes: la presencia de inestabilidad de microsatélites y la pérdida de expresión de la proteína correspondiente al gen afecto (detectable mediante inmunohistoquímica).
Criterios diagnósticos. Los criterios de Amsterdam, descritos en 1991, fueron clave para definir este síndrome, sin embargo son muy estrictos (<40% de las familias los cumplen) e inicialmente no consideraban el CG. Posteriormente, se han establecido los criterios revisados de Bethesda que son probablemente los más utilizados con el objetivo de determinar en qué pacientes con CCR se debería realizar el estudio del sistema reparador del ADN en el tumor, para centrar el análisis genético en línea germinal en aquellos pacientes con alteración de dicho sistema (objetivada por presencia de inestabilidad de microsatélites y/o pérdida de expresión proteica por inmunohistoquímica para MLH1/MSH2/MSH6/PMS2). De forma paralela al desarrollo de criterios clínicos, en los últimos años se han establecido diferentes modelos matemáticos, cuyo objetivo es facilitar la identificación de pacientes con SL mediante la predicción del riesgo de ser portador de una mutación en alguno de los genes reparadores del ADN en base a la historia personal y familiar de cáncer. Los principales modelos actualmente son MMRpro, MMRpredict y PREMM1,2,6. Pese a que estos modelos han demostrado ofrecer una sensibilidad y especificidad satisfactorias, y además cuantificar el riesgo de ser portador de mutación, se necesita de la sospecha clínica y de obtener una historia familiar completa y precisa por lo que actualmente son utilizados principalmente en centros que evalúan pacientes de alto riesgo21,22.
Cribado de CG. No está bien establecido. Algunos grupos recomiendan la determinación y erradicación de H. pylori23.
Pese a que no existe evidencia sólida de la conveniencia de gastroscopia de cribado en los pacientes con SL, el cribado endoscópico se aconseja en países con alta prevalencia de esta neoplasia, así como en familias con SL con al menos un familiar con CG. Periodicidad: cada 1-3 años, a partir de los 30-35 años23.
Síndrome de Li-FraumeniEs un síndrome de baja prevalencia que conlleva un riesgo incrementado de múltiples tumores primarios. El 50% de los individuos portadores de mutación germinal en el gen TP53 desarrollan un cáncer asociado a este síndrome antes de los 30 años. Las 4 neoplasias principales son el cáncer de mama, cerebral, adrenocortical y el sarcoma (representando el 80% de los casos). Dentro de otras neoplasias menos frecuentes se encuentran la leucemia, cáncer de pulmón, melanoma, cáncer pancreático y cáncer gástrico. La frecuencia de CG en este síndrome es de 2,8%24,25.
Genética. Herencia autosómica dominante causada por mutación germinal en el gen TP53 localizado en el cromosoma 17p13. TP53 es un gen supresor de tumores cuya función principal es regular las células con ADN dañado, cuando la proteína p53 funciona de forma incorrecta, las células con ADN dañado pueden sobrevivir y proliferar contribuyendo a la transformación maligna.
Anatomía patológica. La histología tumoral puede ser intestinal (70% de los casos) o difusa (30% de los casos).
Criterios diagnósticos. Una mutación en TP53 se debe sospechar en un individuo con alguno de los siguientes 3 criterios diagnósticos (criterios de Chompret 2009)26:
- •
Individuo con:
- ∘
Tumor del espectro del síndrome diagnosticado a <46 años (sarcoma de tejidos blandos, osteosarcoma, cáncer de mama en mujer premenopáusica, tumor cerebral, carcinoma adrenocortical, leucemia o cáncer broncoalveolar) y,
- ∘
Al menos un FPG o FSG con algún tumor del espectro del síndrome (excepto cáncer de mama si el probando ha tenido un cáncer de mama) <56 años de edad o con múltiples tumores.
- ∘
- •
Individuo con múltiples tumores (excepto tumores múltiples de mama), 2 de los cuales sean del espectro del síndrome, alguno diagnosticado a <46 años.
- •
Individuo con un carcinoma adrenocortical o tumor de plexos coroides, independientemente de la historia familiar.
Cribado de CG. Está indicado si hay casos de CG en la familia. Periodicidad: cada 2-3 años, la edad de inicio se establece en base a la edad de presentación del CG en el familiar afecto27.
Síndrome de Peutz-JeghersEs un síndrome que se caracteriza por la presencia de pólipos hamartomatosos a lo largo del tracto digestivo e hiperpigmentación mucocutánea. El riesgo acumulado de desarrollar un cáncer a los 70 años es de 85-90%. Las neoplasias más comunes son el cáncer de mama y de colon, seguidos del cáncer de páncreas, estómago y ovario. El riesgo estimado de CG es de 30% con una edad media al diagnóstico de 30-40 años de edad28,29.
Genética. Herencia autosómica dominante. Se identifica en el 70% de los casos una mutación germinal causal en el gen STK11 (también conocido como LKB1) localizado en el cromosoma 19p13.3. STK11 es un gen supresor de tumores que aparentemente regula la polaridad celular, lo que tiene relación con el desarrollo de hamartomas y el riesgo incrementado de cáncer30.
Anatomía patológica. La histología tumoral predominante es de adenocarcinoma intestinal.
Criterios diagnósticos. El diagnóstico clínico se establece con la presencia de dos o más de los siguientes criterios:
- •
Pigmentación mucocutánea.
- •
Dos o más hamartomas gastrointestinales tipo Peutz-Jeghers.
- •
Historia familiar de SPJ.
Cribado de CG. Se recomienda una exploración basal a los 8 años y continuar cada 3 años. Si no hay pólipos, reiniciar el cribado endoscópico a los 18 años, cada 3 años31.
Poliposis adenomatosa familiarEs un síndrome caracterizado por la presencia de múltiples adenomas colorrectales, que en su forma clásica (más de 100 adenomas) tiene un riesgo casi del 100% de presentar cáncer colorrectal a edad temprana si no se realiza colectomía profiláctica. Se asocia a un amplio espectro de tumores extracolónicos entre los que destacan el hepatoblastoma, adenocarcinoma duodenal, pancreático, tiroideo, de vía biliar y cerebral. El riesgo de CG estimado es de 0,5-2%32.
Genética. Herencia autosómica dominante. Asociado a mutaciones germinales en el gen APC (cromosoma 5q21-q22) en más del 50% de los casos de poliposis con más de 100 adenomas. El gen codifica una proteína de 2,843 aminoácidos (310kDa) que interviene en la vía de señalización Wnt. APC es un gen supresor de tumores que funciona regulando de forma negativa la oncoproteína β-catenina que participa en la regulación de la transcripción de genes como proliferación, diferenciación, apoptosis, etc.33. El tipo de mutación más frecuente es la que trunca la proteína, aunque puede haber deleciones exónicas y de genes completos.
Anatomía patológica. El CG en este síndrome es de tipo intestinal.
Criterios diagnósticos. La sospecha diagnóstica se basa en dos fenotipos principales, la forma clásica, caracterizada por más de 100 adenomas a lo largo de todo el colon y la forma atenuada que presenta entre 10 y 99 adenomas.
Cribado de CG. Reservado para aquellos pacientes con historia familiar de CG, se recomienda gastroscopia cada 3-5 años, la edad de inicio no está establecida y se decide en función de los antecedentes familiares. En la mayoría de los casos, la estrategia de vigilancia con gastroscopia está establecida en base al seguimiento de los pólipos duodenales, realizando una primera exploración a los 25-30 años. Los pólipos gástricos más frecuentes en la PAF son los pólipos de glándulas fúndicas, y en menor frecuencia los adenomas. Dada la baja prevalencia de CG en la PAF, solo está indicada la toma de biopsias/polipectomía de aquellos pólipos gástricos que muestren cambios sugestivos de malignidad, especialmente los localizados en antro34,35.
Poliposis asociada a MUTYHEs un síndrome caracterizado por un fenotipo de poliposis adenomatosa atenuada, con la presencia de menos de 100 adenomas y en algunos casos manifestaciones extracolónicas similares a la PAF. El riesgo de neoplasias extracolónicas parece ser menor que en la PAF. El riesgo de CG es del 1%.
Genética. Es un síndrome autosómico recesivo, causado por mutaciones germinales bialélicas en el gen MUTYH (cromosoma 1p34.3–1p32.1). MUTYH es un gen de reparación por escisión de bases cuyas proteínas son las encargadas de reparar el daño oxidativo del ADN. Los genes mutados como consecuencia del daño oxidativo ejercen una influencia importante sobre el fenotipo de la poliposis y la inactivación de MUTYH genera una inestabilidad genómica en las células del epitelio colorrectal incrementando de esta manera el riesgo de cáncer colorrectal36. La mayoría de las mutaciones son mutaciones de cambio de aminoácido (missense). Existen diversos estudios que apoyan la variabilidad de las mutaciones en MUTYH en base a las diferencias geográficas y étnicas. Así, en nuestro país, las dos mutaciones más prevalentes son G396D y Y179C (que corresponden a las más frecuentes en población caucásica), aunque también se han reportado otras mutaciones más específicas de nuestra área geográfica, como p.E410GfsX4337.
Anatomía patológica. No existe información sobre el tipo histológico de CG en este síndrome.
Criterios diagnósticos. Se sospecha en pacientes con fenotipo de poliposis adenomatosa atenuada o clásica con patrón de herencia recesivo.
Cribado de CG. Al igual que en la PAF, el cribado de CG queda reservado para aquellos pacientes con historia familiar de CG, se recomienda gastroscopia cada 3-5 años, la edad de inicio no está establecida y se decide en función de los antecedentes familiares. En la mayoría de los casos, la estrategia de vigilancia con endoscopia alta está establecida en base al seguimiento de los pólipos duodenales, realizando una primera exploración a los 25-30 años38.
Poliposis juvenilEs un síndrome caracterizado por múltiples pólipos hamartomatosos en el tracto digestivo (principalmente en el colon y estómago) y un riesgo aumentado de cáncer gastrointestinal. Es el síndrome hamartomatoso más común, con una incidencia de uno en 100.000 nacidos. El riesgo acumulado de CG es del 21%39.
Genética. Herencia autosómica dominante. En el 50% de los casos se asocia a mutación germinal en el gen SMAD4 localizado en el cromosoma 18q21.1, BMPR1A localizado en el cromosoma 10q22-23 o ENG localizado en el cromosoma 9q34.1. Los tres genes están relacionados con la vía de señalización del factor de crecimiento transformador beta (TGF-beta)40.
Anatomía patológica. La histología tumoral puede ser intestinal o difusa.
Criterios diagnósticos. Se caracteriza por la presencia de al menos uno de los siguientes criterios:
- •
Cinco o más pólipos juveniles en el colon.
- •
Múltiples pólipos juveniles a lo largo del tracto gastrointestinal.
- •
Cualquier número de pólipos juveniles en paciente con historia familiar de poliposis juvenil.
Cribado de CG. Se recomienda realización de gastroscopia cada 1-3 años empezando a los 15 años31.
Síndrome de cáncer de mama y ovario hereditarioEs un trastorno con un riesgo aumentado de cáncer de mama (47-55% a los 70 años), de cáncer de ovario (17-39%) y otros tipos de cáncer, incluyendo próstata, mama masculina, melanoma, cáncer de páncreas y cáncer de estómago. El riesgo de CG es de 2,6-5,5%41. En base a un metaanálisis, se estima que el riesgo relativo de CG es de 1,69 (IC 95%, 1,21-2,38)42.
Genética. Herencia autosómica dominante. La mayoría de los casos se debe a mutaciones en los genes BRCA1 (17q21.31) o BRCA2 (13q13.1) y menos frecuentemente en el gen PALB2 (16p12.2)43,44. BRCA1 y BRCA2 son genes supresores de tumores y tienen un papel principal en la respuesta al estrés celular a través de procesos de reparación del ADN45. El PALB2 es un gen asociado a BRCA2 y tiene una función de supresor tumoral involucrada en mantener la integridad genómica46.
Anatomía patológica. La histología tumoral puede ser intestinal o difusa.
Criterios diagnósticos. Una mutación en BRCA1/BRCA2/PALB2 se debe sospechar en un individuo con alguno de los siguientes criterios diagnósticos:
- •
Cáncer de mama <40 años.
- •
Cáncer de mama y ovario.
- •
Cáncer de mama múltiple (ipsi/contralateral).
- •
Cáncer de mama en sexo masculino.
- •
Cáncer de mama con el receptor de estrógeno, progesterona y HER2/neu negativos («triple negativo»).
- •
Cáncer de mama y páncreas.
- •
Cáncer de ovario seroso papilar de alto grado.
- •
Cáncer de mama a cualquier edad y:
- ∘
Dos o más familiares con cáncer de mama o páncreas (independientemente de la edad).
- ∘
Uno o más familiares con cáncer de mama <50 años.
- ∘
Uno o más familiares con cáncer de ovario (independientemente de la edad).
- ∘
Tres o más familiares con cáncer de mama (independientemente de la edad).
- ∘
Cáncer de páncreas en paciente o familiar con cáncer de mama o de ovario.
- ∘
Cribado de CG. No existe una recomendación sólida. Algunos autores recomiendan la realización de gastroscopia cada 2-3 años en pacientes con historia familiar de CG, sin embargo no hay ningún consenso ni guía que apoye dicha recomendación38.
Cáncer gástrico familiarSe define como aquellos casos con agregación familiar de CG de histología intestinal, sin una causa genética identificada. Se estima que los FPG de pacientes con CG tienen un riesgo incrementado de 2-3 veces de presentar dicha neoplasia, en relación con la población general47.
Genética. Se desconoce la mutación germinal causal.
Criterios diagnósticos. Los criterios que definen al CG familiar son:
> 3 FPG o FSG con CG, con independencia de la edad, o
> 2 FPG o FSG con CG, al menos uno afecto antes de los 50 años.
Anatomía patológica. La histología tumoral es intestinal (ya que si es histología difusa entra dentro de los criterios diagnósticos de CGDH).
Prevención y cribado de CG. No recomendada la gastrectomía profiláctica. Se aconseja la realización de una primera gastroscopia a partir de los 40 años o 5 años antes de la edad del familiar afecto más joven, con detección y erradicación de H. pylori2. No existe consenso en la conveniencia de gastroscopias periódicas y el intervalo entre ellas, recomendando algunos autores una exploración cada 2-5 años en función de la historia familiar (fig. 2)3.
Cáncer pancreáticoEl cáncer de páncreas (adenocarcinoma ductal pancreático [ACDP]) es una enfermedad rápidamente progresiva y generalmente fatal, representando a nivel mundial la octava causa de muerte por cáncer en hombres y la novena en mujeres48. En España, la incidencia es de 6,3 casos por 100.000 habitantes1. La edad media al diagnóstico es de 71 años, tan solo un 8-10% se presentan antes de los 50 años. Aunque los tratamientos han mejorado, el ACDP tiene una supervivencia media a los 5 años de <5%49,50.
El tabaquismo, índice de masa corporal alto, el consumo excesivo de alcohol, y la diabetes mellitus son factores asociados a un mayor riesgo de ACDP. Dentro de los factores de riesgo, el tabaquismo es el factor de riesgo exógeno más estrechamente relacionado con el desarrollo de ACDP. Se asocia a un aumento de riesgo de 2 a 3,7 veces, y disminuye la edad de aparición del cáncer en alrededor de 10 años.
Así mismo, una historia familiar de cáncer pancreático también se ha relacionado con un mayor riesgo de desarrollar esta neoplasia, lo que sugiere un componente hereditario y que los factores genéticos pueden jugar un papel importante en el desarrollo de este tumor. Aproximadamente el 5-10% de los pacientes con ACDP presentan antecedentes familiares de esta neoplasia, observándose por tanto, cierto grado de agregación familiar. Se han identificado varias mutaciones en línea germinal involucradas en el ACDP hereditario51.
Hasta la actualidad no se ha identificado un gen causal principal de ACDP, pero existe una asociación a mutaciones germinales que conlleva un mayor riesgo de esta neoplasia. Además, estudios recientes han descrito el fenómeno de anticipación en el 59-85% de las familias, lo cual implica que las generaciones más jóvenes suelen afectarse 10 años antes52. Las alteraciones genéticas más frecuentes son: BRCA2, PALB2 que se une a la proteína BRCA2, ATM, CDKN2A/p16, y menos frecuentemente: BRCA1, APC, MLH1, MSH2, MSH6, PMS2, PRSS1 y STK1150,53,54.
Existen tres situaciones clínicas en las que se puede encontrar predisposición a ACDP, dos situaciones englobadas dentro del cáncer pancreático hereditario (CPH; mutación germinal asociada) y la tercera definida como cáncer pancreático familiar (CPF; sin mutación genética identificada).
- 1)
CPH en contexto de síndromes hereditarios con mayor riesgo principalmente de cáncer pancreático:
1.1 Pancreatitis hereditaria (PH).
1.2 Fibrosis quística.
- 2)
CPH en contexto de síndromes hereditarios con mayor riesgo de ACDP y otros tumores: en contexto de otros síndromes de cáncer hereditario que asocian un mayor riesgo de ACDP y de otras neoplasias, con una mutación germinal asociada (tabla 1).
- 3)
Cáncer de páncreas familiar: situación en la que se observa una agregación familiar de ACDP, sin una causa genética identificada.
Es una enfermedad poco frecuente, constituyendo una forma hereditaria de pancreatitis crónica. La aparición de los síntomas comienza entre la primera y la segunda década de la vida. Se presentan cuadros recurrentes de pancreatitis aguda que llevan finalmente al desarrollo de pancreatitis crónica, insuficiencia pancreática, diabetes y riesgo de ACDP, el cual oscila entre 18 y 53% en estudios recientes54,55.
Genética. Patrón de herencia autosómico dominante. Se asocia a la mutación en el gen PRSS1 (ubicado en el cromosoma 7q35), con una alta penetrancia (estimada del 80%). PRSS1 codifica la proteína tripsina 1 que es la principal catalizadora de la conversión de zimógenos pancreáticos en enzimas pancreáticas. En algunos casos la PH está asociada al gen SPINK1 (ubicado en el cromosoma 5q32) y que codifica al inhibidor de la tripsina secretora pancreática.
Criterios diagnósticos. El diagnóstico se basa en la historia clínica apoyada por pruebas complementarias de imagen y un patrón de herencia autosómico dominante.
Fibrosis quísticaEs una enfermedad que condiciona una morbimortalidad importante. Se caracteriza principalmente por infecciones respiratorias persistentes, insuficiencia pancreática y niveles elevados de cloro en el sudor. El riesgo de ACDP en estos pacientes no está bien establecido, aunque se considera que es bajo (< 5%).
Genética. Herencia autosómica recesiva. Está causada por mutaciones en el gen CFTR localizado en el cromosoma 7q31.2. CFTR codifica a la proteína reguladora de conductabilidad transmembrana de la fibrosis quística, encargada de regular canales de cloruro.
Criterios diagnósticos:
- •
Síntomas compatibles con fibrosis quística que afecten al menos un órgano y
- •
Disfunción del regulador de conductabilidad transmembrana de la fibrosis quística (CFTR):
- ∘
Cloruro>60mml/l en el sudor, o
- ∘
Identificación de mutación causante de la enfermedad en cada gen CFTR, o
- ∘
Transporte anormal de iones en el epitelio nasal.
- ∘
El CPH puede presentarse en contexto de otros síndromes de predisposición al cáncer, como son el síndrome de CMOH, PAF, SL, SPJ, síndrome de Li-Fraumeni, ataxia telangiectasia y síndrome de melanoma múltiple atípico familiar (FAMMM)54,55.
Dentro de los síndromes hereditarios, las mutaciones de BRCA2 son la forma más común de CPH. El riesgo acumulado de ACDP en contexto del síndrome de cáncer de mama-ovario hereditario es de 3-8%. En España, la prevalencia de mutaciones en PALB2 en familias con HBOC, BRCA1/BRCA2 negativos, e historia personal o familiar de cáncer pancreático es del 1,5%56. Dentro de los síndromes hereditarios que conllevan mayor riesgo de desarrollar esta neoplasia están el SPJ y el de FAMMM (con un riesgo acumulado de 36% y 10-17%, respectivamente). El SPJ a pesar de ser un síndrome raro, confiere actualmente el mayor factor de riesgo hereditario conocido para el ACDP.
CMOH, PAF, SL, SPJ, Sd Li-Fraumeni: ya han sido descritos en el apartado de CG asociado a otros síndromes hereditarios de predisposición a cáncer (tabla 1)Ataxia telangiectasiaEs una enfermedad asociada a un defecto de los mecanismos de reparación del ADN. Los pacientes con dicha patología presentan varias alteraciones neurológicas como ataxia cerebelosa progresiva y movimientos oculares anormales, además de telangiectasias oculares y cutáneas, y deficiencia inmunológica. Dicha enfermedad tiene una herencia autosómica recesiva (mutación bialélica en el gen ATM), sin embargo el riesgo incrementado de ACDP está relacionado a mutaciones en heterocigosis del gen ATM. El riesgo de ACDP en estos pacientes es de al menos el doble que la población general, representando un 2,4% de los pacientes con CPF, porcentaje que aumenta al 4,6% si hay más de tres casos de ACDP en la familia57.
Genética. Mutación del gen ATM (ataxia-telangiectasia mutated gene) localizado en el cromosoma 11q2258. ATM está involucrado en la detección de daño a nivel del ADN y en la progresión del ciclo celular59.
Criterios diagnósticos. El diagnóstico de ataxia telangiectasia se basa en un cuadro clínico compatible y la identificación de la mutación en ambos alelos del gen ATM60, sin embargo como se mencionó anteriormente, el riesgo de ACDP no requiere mutación bialélica61.
Síndrome de melanoma múltiple atípico familiarEs un síndrome asociado a la mutación de un gen supresor de tumores (p16/CDKN2A), que se caracteriza por el desarrollo de múltiples nevus displásicos y melanoma. Los pacientes con dicho síndrome presentan un riesgo incrementado de 13-22 veces de cáncer de páncreas en comparación con la población general62. Un estudio reciente en población española observó una mayor prevalencia de cáncer pancreático (tasa de prevalencia 2,97,p=0,006) en los pacientes con melanoma múltiple portadores de CDKN2A en comparación con los pacientes con melanoma múltiple sin mutación identificada63.
Genética. Herencia autosómica dominante con penetrancia incompleta. Asociado a mutaciones germinales del gen p16/CDKN2A localizado en el cromosoma 9p21. CDKN2A codifica a dos proteínas: p16 y p14ARF. La proteína p16 es un regulador negativo de la progresión del ciclo celular64.
Criterios diagnósticos. La incidencia de mutaciones en CDKN2A es de hecho mayor en los individuos con tres o más melanomas y/o en familias con al menos un miembro con melanoma y otros dos o más familiares de primer o segundo grado con diagnóstico de ACDP.
Los criterios clínicos diagnósticos de FAMMM son:
- •
Melanoma maligno en >1 FPG y presencia de >50 nevus y nevus con características histológicas atípicas.
Representa el síndrome más frecuente de ACDP, en el cual se observa una agregación familiar de esta neoplasia sin una causa genética identificada. El incremento del riesgo de desarrollar esta neoplasia está condicionado por el número de familiares afectos (tabla 2)65.
Genética. Se desconoce la mutación germinal causal.
Criterios diagnósticos. Los criterios que definen al CP familiar son:
> 2 FPG con independencia de la edad, o
> 3 FPG o FSG o FTG con independencia de la edad.
Cribado del adenocarcinoma ductal pancreático hereditario y/o familiarLos individuos con historia familiar de ACDP se consideran de alto riesgo para desarrollar cáncer y por tanto es aconsejable incluirlos en programas de cribado bajo un equipo multidisciplinario. El objetivo del mismo es detectar las lesiones precursoras de ACDP o un cáncer en estadio precoz (tumor papilar mucinoso intraductal [TPMI], neoplasia pancreática intraepitelial grado 3 [PanIN3], T1N0M0)66.
Los TPMI son neoplasias epiteliales intraductales potencialmente malignas, compuestas por células columnares productoras de mucina, con proliferación papilar, formaciones quísticas y grados variables de atipia celular, estas lesiones pueden afectar tanto al conducto principal del páncreas como a las ramas secundarias. Debido al riesgo de malignizar (70% en TPMI de ducto principal) estas lesiones deben ser diagnosticadas y tratadas de forma apropiada67.
Pese a que no existe evidencia sólida de que el cribado se asocie a una disminución de la mortalidad por ACDP, se recomienda incluir en programas de cribado aquellos pacientes que serán candidatos a cirugía si en el estudio se encuentra una lesión sospechosa.
Se recomienda cribado en los siguientes casos68:
- •
Miembros de familias con CPH con mutación germinal conocida:
- a)
Individuos con diagnóstico de SPJ o pancreatitis hereditaria.
- b)
Portadores de mutación en p16/CDKN2A, BRCA1, BRCA2, ATM, PALB2, MSH2, MLH1, MSH6, PMS2, APC, con historia familiar de ACDP.
- a)
- •
Miembros de familias con CPF: individuos con 2 o más familiares de primer grado o individuos con 3 o más familiares (de primero, segundo o tercer grado) con ACDP, siendo al menos uno de ellos familiar de primer grado.
Actualmente la ecoendoscopia y la resonancia magnética son las pruebas de elección para los programas de cribado, ya que son los procedimientos que han demostrado una mayor detección de lesiones precursoras de ACDP. La primera opción es la ecoendoscopia (dada su habilidad para detectar lesiones menores de 10mm) y como alternativa se recomienda la resonancia magnética66,69.
Habitualmente los programas de cribado comienzan a la edad de 40 años o 10 años antes de la edad del familiar afecto más joven. Sin embargo, dos estudios recientes66,70 han demostrado que el rendimiento diagnóstico es significativamente más alto a partir de los 65 años (35 vs.3%), por lo tanto, parece estar justificado aumentar la edad para la primera exploración a los 50 años, a excepción de que los familiares afectos hayan desarrollado el cáncer a una edad temprana. Existen dos situaciones que conllevan mayor riesgo de cáncer precoz y por tanto la estrategia de cribado es diferente: a) en pacientes con SPJ se recomienda iniciar el cribado del ACDP a la edad de 30 años, b) en pacientes con PH se recomienda iniciar el cribado del ACDP a la edad de 40 años. En cuanto a la periodicidad en todos los casos, la recomendación es realizar la exploración anualmente.
Si hay una lesión sospechosa, no hay consenso entre pancreatectomía parcial o total. Una alternativa es la resección parcial y si la anatomía patológica intraoperatoria confirma un ACDP, PanIN2/3 multifocal o TPMI de alto grado de displasia, proceder a la resección pancreática total. La decisión del tipo de intervención quirúrgica ha de individualizarse, tomando en consideración la presencia de insuficiencia pancreática exocrina y endocrina. En pacientes asintomáticos, no está indicada la pancreatectomía total profiláctica55,66 (fig. 3).
Perspectiva en el futuro
La causa genética subyacente para un alto porcentaje de familias con agregación de CG y CP no ha sido identificada. La dificultad de establecer criterios diagnósticos para estas familias, asociado al hecho de que un mismo fenotipo puede ser causado por diversos genes y por el contrario un gen puede ser responsable de diversos fenotipos, convierten en un reto su identificación en la práctica clínica.
Por ejemplo, un estudio reciente6 identificó que pacientes que cumplían criterios clínicos de CGDH, con estudio genético en CDH1 negativo, presentaron mutaciones en otros genes (CTNNA1, BRCA2, STK11, entre otros) sugiriendo que el síndrome de CGDH se puede definir por mutaciones en CDH1 así como en otros genes relacionados.
Actualmente con la disponibilidad de la tecnología de secuenciación de nueva generación, probablemente se descubran nuevas mutaciones genéticas responsables de cáncer gástrico y pancreático hereditarios. Así mismo, con la pronta llegada de paneles de secuenciación de nueva generación a nivel comercial, la aproximación de los estudios genéticos y la complejidad del consejo genético cambiarán de forma significativa. El uso de los paneles multigen permitirá el análisis de diversos genes al mismo tiempo y a un menor coste. Uno de los principales retos que esta nueva tecnología traerá será la interpretación de la información y el manejo de numerosas variantes de significado incierto, siendo necesaria una mayor colaboración multidisciplinar.
Con estas nuevas herramientas diagnósticas, es previsible que en los próximos años se puedan identificar y caracterizar mejor a este subgrupo de pacientes con alto riesgo de CG y CP, y por tanto, profundizar en la patogénesis y comportamiento de esta neoplasia, para poder ofrecer el mejor manejo disponible (estrategias de cribado, vigilancia y tratamiento) con el fin último de reducir la incidencia y mortalidad por cáncer.
ConclusiónLos diferentes síndromes que predisponen al cáncer gástrico hereditario y/o cáncer pancreático hereditario son patologías poco frecuentes pero con una morbimortalidad importante. Varios de ellos están asociados a mutaciones conocidas, pero en la mayoría no es posible identificar una causa genética. En función del síndrome y/o de la historia familiar, se establecen criterios de diagnóstico precoz y vigilancia, de cara a intentar disminuir la morbimortalidad ocasionada por estas neoplasias. Por estas razones, la vigilancia de familiares de alto riesgo de estas patologías se debe realizar en centros especializados y si es posible dentro de un protocolo de investigación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

