




INTRODUCCION
La historia del trasplante hepático (TH) se remonta al año de 1963, cuando Thomas Starzl realizó el primer TH en un niño que padecía atresia biliar, el cual sólo sobrevivió 5 h. En el mismo año, 2 meses más tarde, Starzl practicó su segundo TH, esta vez en adultos, y se considera a éste como el primer TH exitoso de la historia. El paciente falleció a los 22 días del postoperatorio a causa de una embolia pulmonar.
El TH está indicado en las enfermedades hepáticas progresivas en las que no sean posibles otras medidas terapéuticas y en las que la supervivencia esperada al año sea inferior a la que se conseguiría con el trasplante1. Las indicaciones más comunes para el TH son la cirrosis secundaria a infección crónica por el virus de la hepatitis C y la cirrosis alcohólica. En niños, la indicación principal suele ser la atresia biliar. Otras enfermedades hepáticas susceptibles de ser tratadas con TH comprenden cirrosis causada por el virus de la hepatitis B, hepatitis autoinmune, colangitis esclerosante primaria, carcinoma hepatocelular, enfermedades metabólicas (como la enfermedad de Wilson o la hemocromatosis hereditaria), e insuficiencia hepática aguda y grave; las causas comunes son las reacciones por hipersensibilidad o idiosincrasia a fármacos, entre otras1,2.
Si bien el TH es la mejor opción terapéutica para determinadas entidades patológicas, como las mencionadas anteriormente, la isquemia-reperfusión (I/R), inherente a todo TH, es la causa principal tanto del mal funcionamiento inicial como del fallo primario del injerto hepático3-5; este último es responsable del 81% de los retrasplantes durante la primera semana tras la intervención quirúrgica6. Y si esto ocurre en hígados sanos, aún son mayores los casos de disfunción primaria cuando el injerto es esteatósico, de ahí que la esteatosis sea la causa del mayor número de óganos no aptos para trasplante (54%), acentuando así la problemática del banco de órganos7. Por tanto, minimizar los efectos adversos del síndrome de I/R podría aumentar el número de órganos disponibles para transplante y el de pacientes que se recuperen exitosamente de un trasplante hepático. El primer paso para lograr este objetivo sería el completo entendimiento de los mecanismos involucrados en la lesión por I/R.
FISIOPATOLOGIA DE LA LESION POR ISQUEMIA-REPERFUSION
Si bien la lesión por I/R se ha dividido en 2 fases, la lesión causada por la isquemia y la lesión debida a la reperfusión, la separación de los episodios celulares que ocurren en cada fase no es absoluta, ya que el daño celular en el órgano hipóxico se acentúa después de la restauración del aporte de oxígeno8, lo que sugiere que los episodios que suceden en la reperfusión son la consecuencia de los que se inician durante la isquemia. Además, se ha observado que la reperfusión de un hígado expuesto a un período de isquemia breve no induce ningún daño detectable, un hallazgo indicativo de que la reperfusión por sí sola no es perjudicial9-11.
Durante la isquemia, se interrumpe el aporte de oxígeno al órgano y se detiene la cadena respiratoria mitocondrial, lo que comporta una depleción en los valores de adenosine triphosphate (ATP)12. La degradación de ATP estimula la glucólisis anaeróbica con la consiguiente formación de ácido láctico. La acidosis resultante, además de alterar la cinética normal de las enzimas, supone un sistema menos efectivo para producir ATP y las células se ven privadas de la energía necesaria para mantener la homeostasis. El fallo en la homeostasis celular se caracteriza por la pérdida de gradiente de los iones de sodio y de calcio a través de las membranas celulares4,5,13. Este hecho provoca un edema intracelular y el consiguiente hinchamiento de las células de Kupffer (CK) y las células endoteliales (CE)14. Estos fenómenos inducen una alteración en los orgánulos citoplasmáticos y en la integridad de la membrana, y pueden desencadenar la muerte celular13. El hígado esteatósico es más vulnerable a los efectos nocivos derivados de la isquemia; en este sentido, numerosos estudios evidencian que en presencia de esteatosis los valores de ATP son significativamente más bajos comparados con hígados sanos7.
La reperfusión (recuperación del flujo sanguíneo) del hígado previamente isquémico inicia toda una serie de fenómenos inflamatorios en los que están implicados múltiples mediadores de la inflamación, plaquetas, leucocitos y el endotelio vascular, los cuales, al interaccionar, derivan en la lesión por reperfusión15,16. A título de ejemplo, entre los mediadores inflamatorios descritos en la lesión por I/R hepática destacan los radicales libres de oxígeno (RLO)17, interferón beta (IFN-ß)18, interferón gamma (IFN-*), tumor necrosis factor beta (TNF-ß), granulocyte-macrophage colony-stimulating factor (GM-CSF)19, tumor necrosis factor alpha (TNF-*), interleucina (IL)-1, IL-1220, IL-1821, leukotriene B4 (LTB4), 12-hydroxyeicosatetraenoic acid (12-HETE)22 y platelet activating factor (PAF)19. Además, también se ha demostrado la participación de IL con propiedades antiinflamatorias que funcionan como reguladores del proceso inflamatorio que se desarrolla en la I/R, como IL-6, IL-1319 y IL-1020. Los mediadores inflamatorios son modulados a nivel transcripcional. Estudios sobre transducción de señales en I/R hepática han descrito un papel notorio para diferentes factores de transcripción como nuclear factor kappa B (NF-*B), activator protein 1 (AP-1)17, peroxisome proliferator-activated receptor-alpha (PPAR-*)23, factor nuclear high mobility group box-1 (HMGB1)24, signal transducer and activator of transcription (STAT3)25, signal transducer and activator of transcription 6 (STAT6)19, hipoxia-inducible factor 1 complex (HIF-1) y heat shock factor (HSF)17. La lesión por reperfusión también implica a las cinasas intracelulares que activan factores de transcripción, como SAPK, c-Jun N-terminal kinase (JNK) o p38 mitogen-activated protein kinase (p38 MAPK)17.
Respecto a la acumulación de neutrófilos, se considera que este proceso está regulado por la participación de citocinas, factores del complemento, moléculas de adhesión y quimiocinas22, que permiten el reclutamiento, la adhesión y la transmigración de neutrófilos. Algunas moléculas de adhesión celular, conocidas por su papel en I/R hepática, comprenden E-selectina, P-selectina19, L-selectina16, integrinas ß1, integrinas ß2, intercellular adhesion molecule-1 (ICAM-1) y vascular cell adhesion molecule-1 (VCAM-1)22. De la misma manera, se ha sugerido la participación de quimiocinas, entre las que se han mencionado la IL-8 y sus homólogos20, el cytokine-induced neutrophil chemoattractant-1 (CINC)19, la epithelial neutrophil activating protein-78 (ENA-78)8, la macrophage inflammatory protein-1 (MIP-1) y 28, la keratinocyte-derived chemokine (KC)19 y el monocyte chemoattractant-1 (MCP-1), 2, y 38.
Por último, la disfunción microcirculatoria se origina a partir de una serie de episodios que involucra la interacción de células intravasculares (p. ej., neutrófilos) con células no parenquimales (como las CE y las CK) y que es mediada por la síntesis y liberación de moléculas de adhesión, citocinas, factores del complemento, RLO, óxido nítrico (NO) y endotelinas (ET)26,27.
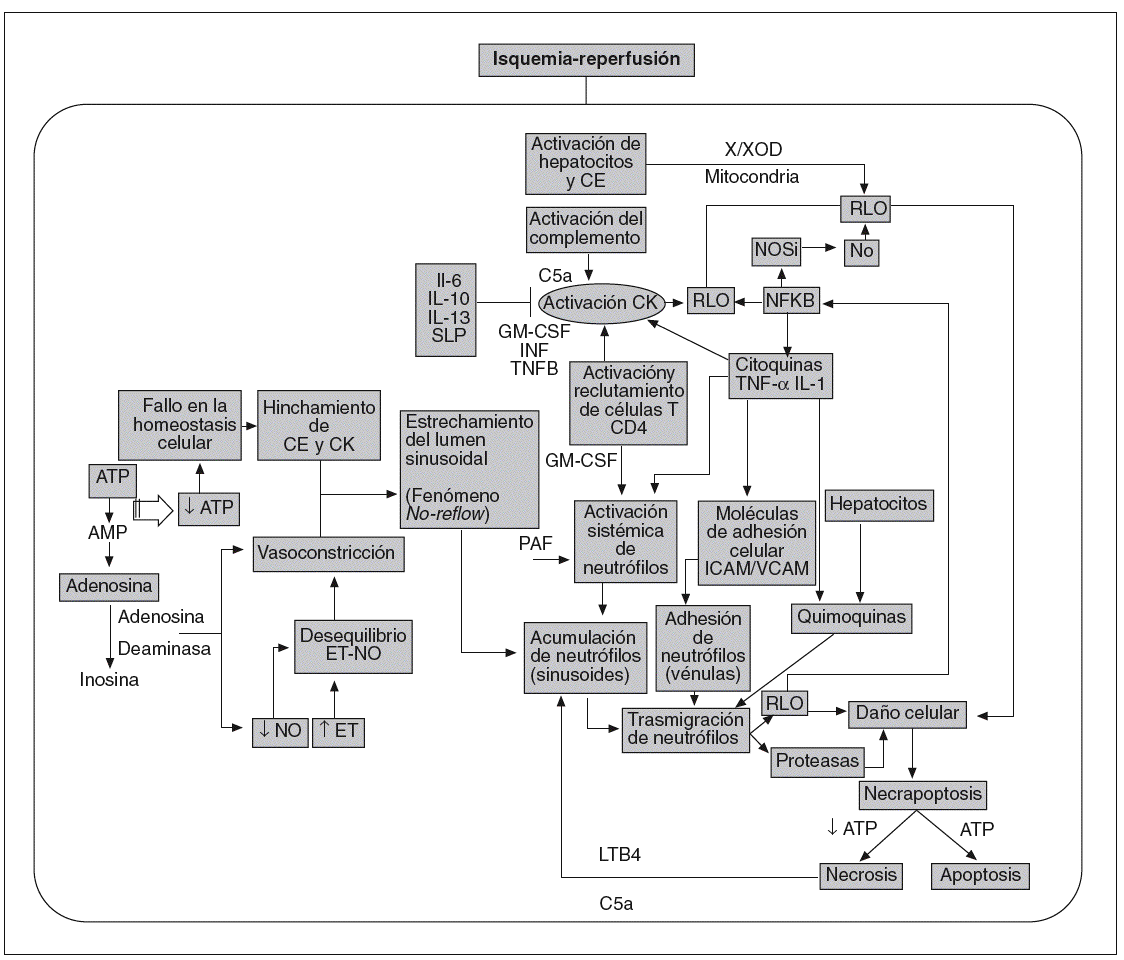
Lo anteriormente expuesto evidencia la multitud de mediadores y factores implicados en la lesión por I/R hepática. Ilustrando lo anterior, en la figura 1 se indican algunos de los mecanismos involucrados. Además, las interrelaciones entre las vías de señalización participantes son muy complejas y aún no es posible hablar con total certeza de los episodios que suceden desde que se inicia la reperfusión hasta que tiene lugar la disfunción o el fallo primario del injerto hepático, pues las diversas investigaciones que abordan el tema no han logrado converger en sus resultados, tal vez en parte por la gran diversidad de modelos y diseños experimentales con los que se trabaja. En esta revisión nos centraremos en los diferentes resultados recogidos en la bibliografía acerca de las po sibles fuentes generadoras de RLO, de los efectos y mecanismos de acción del NO, del papel de determinados mediadores proinflamatorios, como el TNF, y de determinados factores de transcripción como el NF-*B. Estos datos nos permitirán entender aún más el porqué la I/R hepática continúa siendo un problema sin resolver en la práctica clínica.
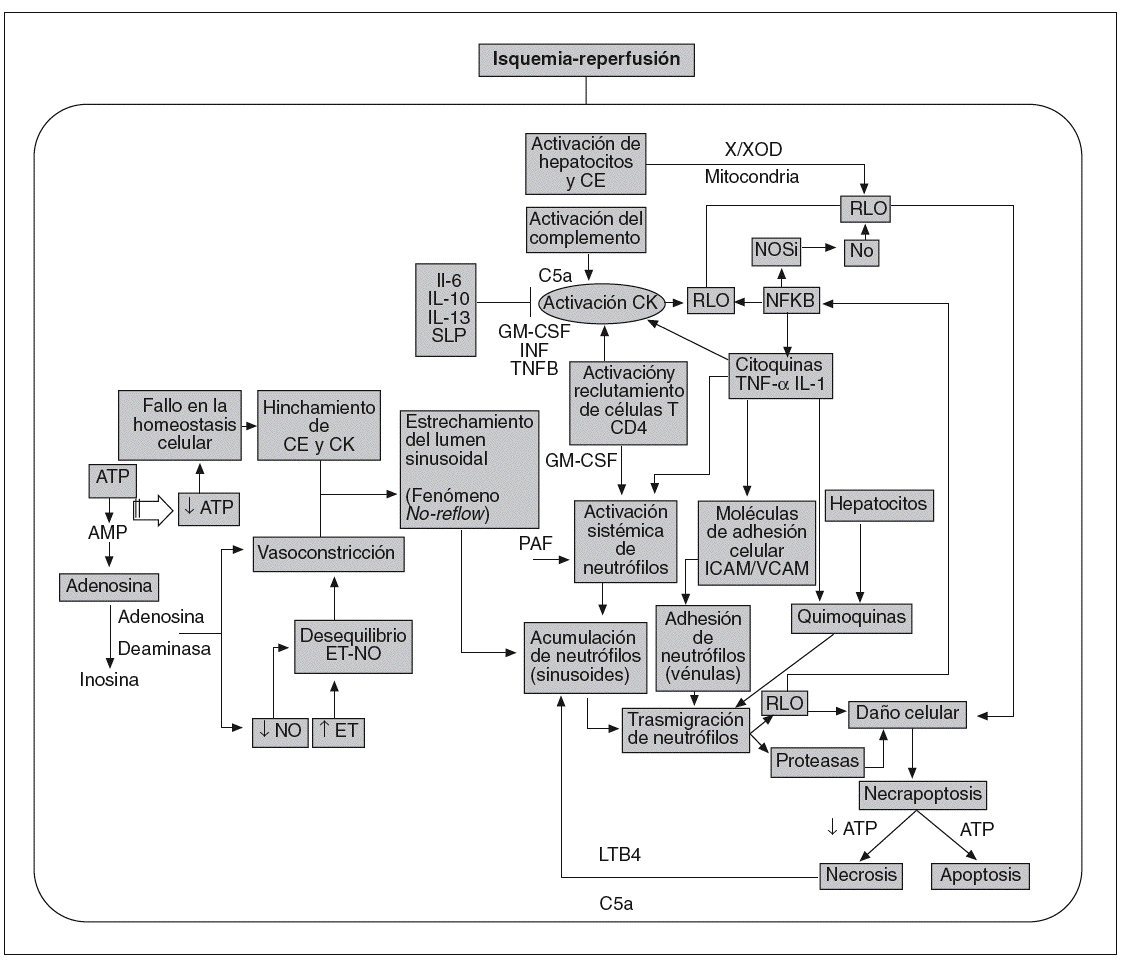
Fig. 1. Mecanismos involucrados en la patofisiología del síndrome por isquemia-reperfusión.
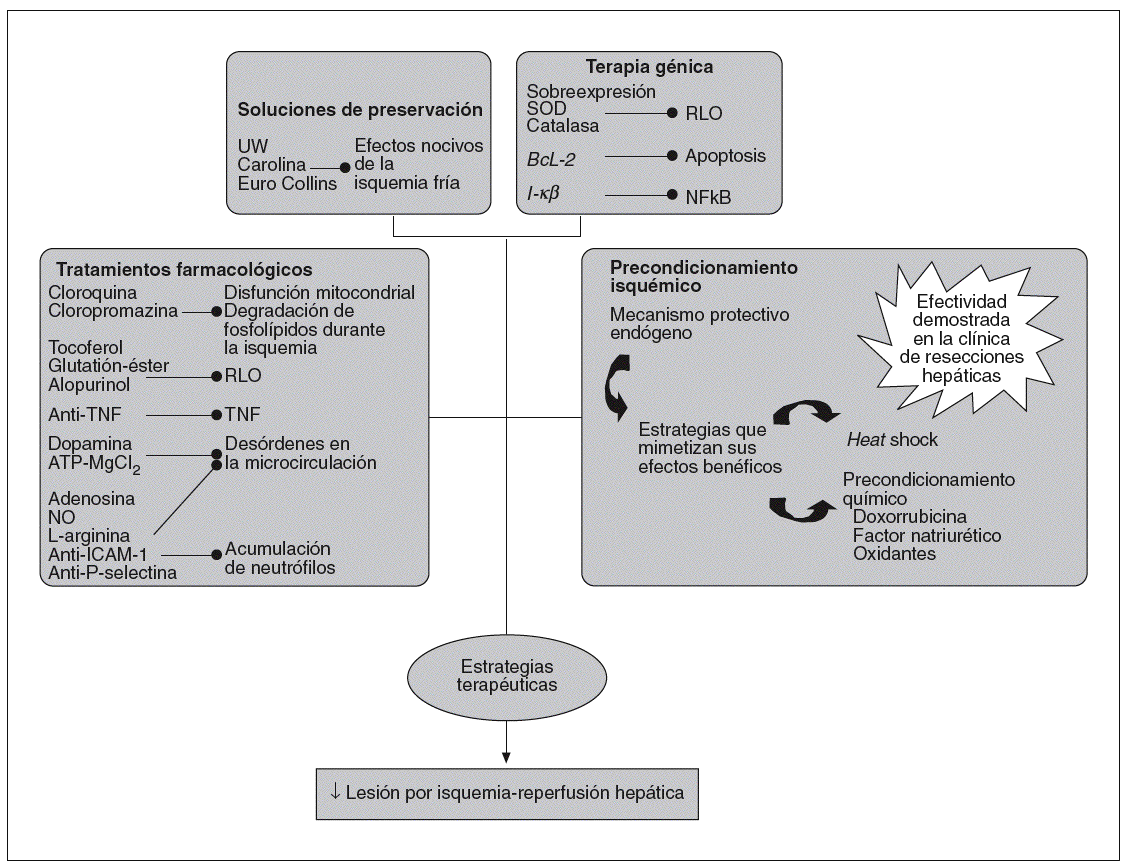
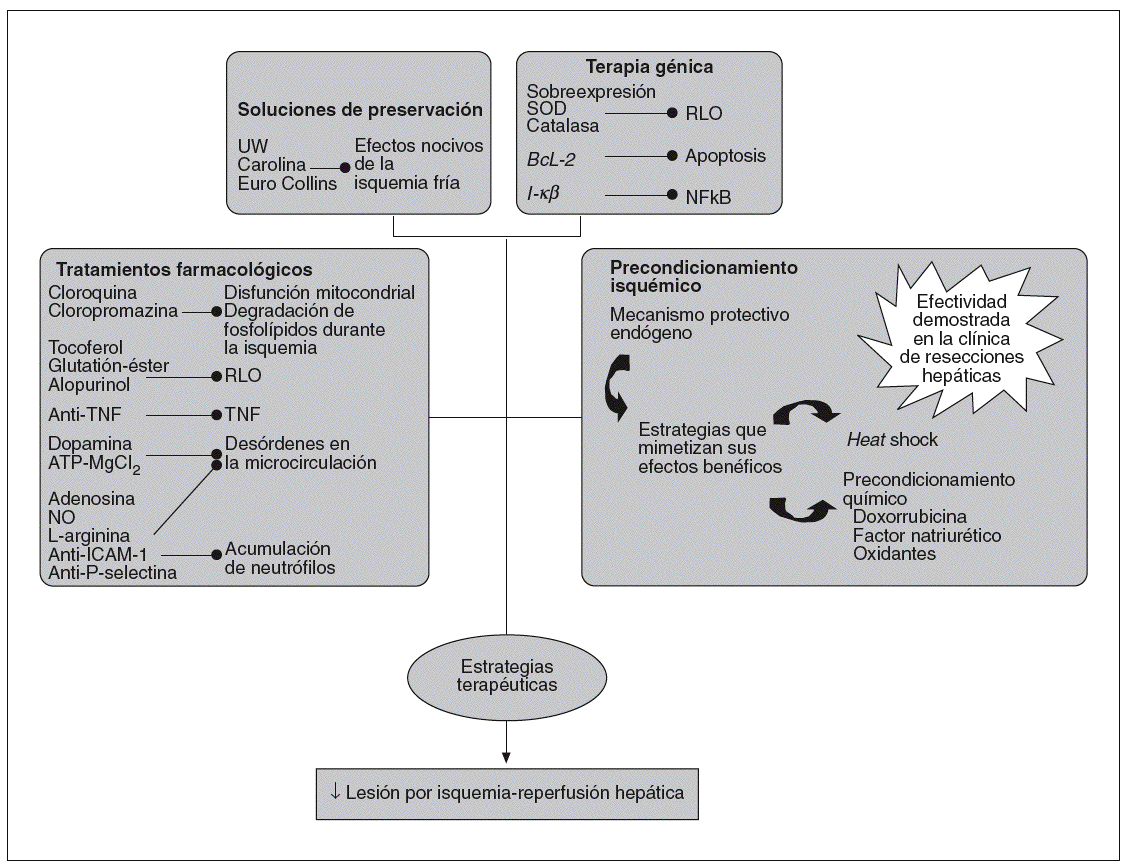
Fig. 2. Estrategias terapéuticas utilizadas para la prevención del daño causado por el síndrome de isquemia-reperfusión.
Respecto a los mecanismos responsables de RLO, los estudios realizados con inhibidores del sistema xantina/xantina oxidasa (X/XOD)28 apuntaban a este sistema como el principal generador de RLO en hepatocitos29,30, implicándolo además en el daño pulmonar asociado al TH31. Por otro lado, otros resultados obtenidos en modelos experimentales de hígado perfundido aislado e isquemia caliente han puesto en tela de juicio la importancia del sistema X/XOD, e indican que la mitocondria podría ser la fuente principal de RLO32. De todas maneras, los datos obtenidos en un modelo murino de I/R caliente in vivo constatan que el papel de la mitocondria no es absoluto33. En la misma línea, otras investigaciones señalan que son los neutrófilos o las CK las principales fuentes generadoras de RLO34,35. Sin embargo, esta última hipótesis también ha suscitado cierta controversia. La eliminación de las CK en TH no modificó los efectos nocivos de la I/R36, y los neutrófilos activados no son esenciales en el daño por reoxigenación8. La divergencia de resultados respecto a los mecanismos de generación de RLO en la I/R hepática es clara. Hay que destacar además que el estrés oxidativo en hepatocitos y la estimulación de CK después de la I/R depende del tiempo de isquemia al cual esté sometido el hígado, y también puede diferir entre isquemia a 4 y 37 ºC37, probablemente conduciendo a diferentes mecanismos de desarrollo del daño hepático. Además, cabe destacar que en la lesión por reperfusión es difícil establecer una distinción entre los mediadores que tienen un papel beneficioso en la lesión por I/R y los que causan efectos perjudiciales. Si bien numerosos estudios destacan el papel protector del NO para reducir la lesión y aumentar la supervivencia en procesos de I/R hepática38-40, los resultados obtenidos por otros autores no destacan ningún efecto del NO, e incluso refieren una acción perjudicial de este mediador vasoactivo en el daño por I/R hepática41. Además, se han encontrado diferencias en los mecanismos de acción del NO, en función de si éste es endógeno o exógeno. En este sentido, mientras que el NO endógeno puede reducir la acumulación de neutrófilos, la suplementación exógena de NO no modifica este parámetro pero se asocia con una inhibición en la producción de ET42. Se habla además de un efecto dual del NO en la I/R hepática, en función de si el NO deriva de la óxido nítrico sintasa constitutiva (NOSc) o de la inducible (NOSi). Si bien la producción de NO derivada de la NOSc tendría efectos benéficos en la I/R hepática, se ha señalado que la producción de NO derivada de NOSi podría contribuir al daño hepático43. En este sentido, hay estudios que proponen que el NO derivado de la NOSi participa en la generación de RLO e induce la liberación de citocromo C y de caspasas, favoreciendo los procesos proapoptóticos44,45. Aún en este punto la controversia continúa, pues otros investigadores han sugerido que la actividad de NOSi es beneficiosa porque induce la expresión de la proteína antiapoptótica Bcl-2 e inactiva las caspasas46,47. Estos efectos diferenciales del NO se han constatado además con otros mediadores implicados en la lesión por I/R hepática. En el caso del TNF, si bien está bien establecido su papel perjudicial en la lesión por I/R hepática, tanto local como sistémica48, se ha observado que este mediador es vital en procesos de regeneración hepática49, presente en el TH con injertos de tamaño reducido (trasplante pediátrico y de donante vivo). Además, se ha visto recientemente en un modelo experimental de I/R hepática que la suplementación de TNF a dosis bajas es altamente eficaz frente al daño por I/R hepática49. Estos efectos diferenciales encontrados en los mediadores inflamatorios se pueden extrapolar además a los factores de transcripción. Es bien conocido que un aspecto común entre la mayoría de los mediadores implicados en la lesión por I/R hepática es su regulación transcripcional. En el caso del NF-*B, experimentos que bloquean la activación de NF-*B en I/R hepática conllevan una reducción en la acumulación de neutrófilos y en el daño hepatocelular50. Estos datos indican que el NF-*B puede ser un componente integral de la respuesta hepática inflamatoria. Sin embargo, otros resultados obtenidos en procesos de I/R hepática ponen de manifiesto que la activación de NF-*B es primordial en la regeneración hepática después de TH51 y reduce la apoptosis y el daño asociados a la I/R hepática.
Se han encontrado también diferentes resultados respecto a la acumulación de neutrófilos. La teoría clásica propone que el aumento en la expresión de moléculas de adhesión, como ICAM-1 y P-selectina, son primordiales para la acumulación de neutrófilos y el consiguiente daño hepático asociado a la I/R16,27. Oponiéndose a esta teoría, también hay resultados que demuestran que la acumulación de neutrófilos en el hígado no depende de la sobrerregulación de ICAM-152. En la misma línea, si bien un aumento en la expresión de P-selectina es importante para desencadenar la lesión pulmonar asociada a la I/R hepática, esta molécula de adhesión, al igual que el ICAM, no participa en la acumulación de neutrófilos en el hígado53. Estos hallazgos apoyan la hipótesis de que durante la I/R hepática, los neutrófilos se acumulan en los sinusoides52, sin depender de las moléculas de adhesión54,55, y tal vez sean los factores mecánicos y las características de la vasculatura hepática los que expliquen este hecho56.
Además de los diferentes resultados encontrados en la bibliografía sobre los mecanismos, las posibles fuentes de mediadores inflamatorios implicados en la lesión por I/R hepática, y de los resultados diferenciales en cuanto a la acción de estos mediadores, también hay una cierta controversia sobre si la muerte celular asociada a la I/R hepática es por necrosis o por apoptosis. Si bien durante mucho tiempo se ha asumido implícitamente que la muerte celular necrótica y apoptótica son entidades diferentes, esta suposición puede no ser válida, lo que ha originado la aparición de un nuevo concepto llamado «necrapoptosis»57.
ESTRATEGIAS PARA PREVENIR LA LESION POR ISQUEMIA-REPERFUSION ASOCIADA AL TRASPLANTE HEPATICO
A pesar de los avances en los tratamientos farmacológicos, en las soluciones de preservación, y en estrategias de terapia génica que han tenido como objetivo disminuir la lesión por I/R asociada al trasplante hepático, los resultados hasta el momento no han sido concluyentes. En la figura 2 se presentan algunas estrategias terapéuticas desarrolladas para prevenir el daño por I/R. En relación con los tratamientos farmacológicos, hay numerosos estu dios experimentales centrados tanto en inhibir los efectos nocivos de la isquemia como la respuesta inflamatoria asociada a la reperfusión. Con esta finalidad, se han administrado fármacos, como la cloroquina58 o la clorpromazina59,60, para prevenir las disfunciones mitocondriales y la degradación de fosfolípidos durante la isquemia hepática. Para inhibir las acciones de los RLO durante la reperfusión, se ha tratado con antioxidantes como tocoferol61, glutatión-éster (GSH-éster) o alopurinol63 y se han administrado también anticuerpos dirigidos al TNF64 para bloquear las acciones nocivas de esta citocina. Para reducir los trastornos microcirculatorios asociados con la I/R hepática, se han realizado tratamientos con dopamina65 y ATP-MgCl266. Se han utilizado también fármacos como adenosina67, donadores de NO68, L-arginina69 y anticuerpos anti-ICAM-170 y anti-P-selectina52 para bloquear la acumulación de neutrófilos. Por otra parte, ninguno de estos tratamientos ha logrado frenar la lesión por I/R hepática. Hay que tener en cuenta las dificultades de frenar la inflamación asociada a este proceso, debido entre otros factores a los múltiples mediadores y tipos celulares implicados en esta respuesta inflamatoria. Además, hay que considerar las dificultades derivadas del tratamiento farmacológico. En este sentido, el GSH-éster no llega al lugar de acción a concentraciones óptimas ni en el momento adecuado71 La administración de anticuerpos anti-TNF provoca sólo una inactivación parcial de la proteína52. Pequeñas variaciones en la dosis de donadores de NO tienen efectos totalmente opuestos42. Además, no hay que descartar los posibles efectos secundarios derivados de los fármacos, ya que en el caso de la dopamina72, adenosina73 y donadores de NO74 se han descrito efectos nocivos sistémicos. Y si esto ocurre en hígados sanos, aún son mayores los problemas para modular la lesión por I/R en hígados esteatósicos. Este tipo de hígados generan RLO que son insensibles a la acción de antioxidantes como la superóxido dismutasa y la catalasa75. La diferencia en los mecanismos de acción entre hígados sanos y esteatósicos supone que los tratamientos efectivos en hígados sanos pueden no serlo en presencia de esteatosis y, además, la dosis de fármaco administrado puede ser diferente en ambos tipos de hígados. Hallazgos como éste deben tenerse en cuenta en el momento de la aplicación por igual de estrategias farmacológicas en I/R en hígados sanos y esteatósicos, pues los efectos pueden ser muy diferentes en cada caso. A título de ejemplo, la administración de un donador de NO en un modelo de trasplante hepático experimental logró reducir el estrés oxidativo en hígados sanos, mientras que la suplementación de NO, a la misma dosis, aumentó la vulnerabilidad de los injertos esteató sicos al síndrome de I/R76. Por otra parte, también po dría haber fármacos que sólo fueran efectivos en hígados esteatósicos. En I/R asociada al TH, se ha evidenciado en hígados esteatósicos un aumento en la expresión de la proteína desacoplante-2 mitocondrial (UCP2) y una capacidad disminuida de sintetizar ATP en la reperfusión, lo que contribuye a la vulnerabilidad de los hígados esteatósicos al síndrome de I/R77. Así pues, fármacos como la cerulenina, que pueden actuar sobre la UCP2 disminuyendo su expresión, logran aumentar el contenido de ATP en hígados esteatósicos78, pero tal estrategia tal vez no tendría ningún efecto en hígados sanos porque no se presenta sobreexpresión de UCP-279. Resultados similares se han obtenido con la carnitina80,81.
Respecto a los avances en la mejoría de las soluciones de preservación, han ido apareciendo diferentes soluciones (Euro Collins, Carolina, Universidad de Wiscosin [UW]) con la finalidad de mantener al injerto en condiciones óptimas durante la isquemia fría. La más utilizada en la clínica ha sido la UW82. La diferencia de esta solución con las restantes es que no contiene glucosa, lo cual evita la producción de lactato y los problemas de acidosis; en cambio, contiene adenosina, que inhibe la agregación plaquetaria y la acumulación de neutrófilos, y es un sustrato para la síntesis de ATP82 y contiene antioxidantes, como GSH y alopurinol, para evitar los efectos nocivos de los RLO83. Una de las limitaciones de la solución de UW es la presencia de hidroxietilamidón, proagreagante de glóbulos rojos, que puede producir un lavado incompleto del injerto hepático con la consiguiente estasis venosa y respuesta inflamatoria84. Otra de las limitaciones es que muchas de las sustancias que se encuentran en la solución UW (alopurinol, lactobionato) no protegen bien por no estar en concentraciones adecuadas o por tener dificultades para llegar al lugar de acción. De hecho, se han realizado estudios en humanos que indican que a pesar de la presencia de alopurinol en UW, este antioxidante no ha logrado frenar los efectos nocivos del sistema generador de RLO, X/XOD85.
En relación con las estrategias de terapia génica centradas en el estrés oxidativo para reducir la lesión por I/R hepática, se han realizado pretratamientos con superóxido dismutasa y catalasa utilizando como vehículo adenovirus, liposomas o polientilenglicol86,87. Para inhibir la apoptosis, se ha sobreexpresado el gen BcL-2, utilizando principalmente como vehículo adenovirus86. Se ha intentado modular la acción del NF-*B tratando con formas mutantes de I-*B88. Por otra parte, los problemas de la terapia génica pasan por la toxicidad de vectores, por la dificultad en conseguir una expresión óptima de la pro-teína en el momento y lugar adecuado, y por la dificultad de conseguir mutantes adecuados (en el caso de NF-*B), debido a las controversias respecto a la activación de NF-*B89-91.
A partir del momento en que se describió la efectividad del precondicionamiento isquémico, consistente en aplicar al hígado breves períodos de I/R previo a la isquemia sostenida92, se han realizado numerosos trabajos con la finalidad de buscar estrategias que puedan mimetizar sus efectos beneficiosos. Una de estas estrategias es el heat shock, que consiste en inducir un aumento en la temperatura corporal antes de la isquemia hepática93. El precon-dicionamiento químico con dexorrubicina94 factor natriurético95 o con oxidantes96 reduce la lesión hepática en diferentes modelos experimentales de I/R. Sin embargo, las limitaciones de estas estrategias es su posible aplicación clínica, ya sea por la dificultad que ello supondría, por problemas de toxicidad y por los efectos secundarios descritos96-98.
Si bien todas estas estrategias (tratamientos farmacológicos, mejoría de las soluciones de preservación, terapia génica, heat shock, precondicionamiento químico) no han podido tener su proyección en la clínica, las investigaciones acerca de la efectividad del PC en modelos experimentales de I/R hepática han sido la base para que esta estrategia quirúrgica pueda ser aplicada en la clínica para reducir la lesión por I/R hepática normotérmica asociada con las resecciones hepáticas de tumores, tanto en hígados sanos como en esteatósicos. Si bien los resultados sobre la efectividad del PC en la clínica fue demostrada por primera vez por Clavien99,100, serán necesarias futuras investigaciones para establecer si esta estrategia quirúrgica puede ser igualmente útil en el TH.
AGRADECIMIENTOS
Este manuscrito ha sido posible gracias a la concesión de los proyectos SAF 2005-00385 y BFI 2003-00912 y al contrato Ramón y Cajal (Dra. C. Peralta) concedidos por el Ministerio de Educación y Ciencia (Madrid). Asimismo, los autores agradecen al Ministerio de Sanidad y Consumo el apoyo recibido (proyecto V2003-REDC03G-O).

