




INTRODUCCIÓN
Los pacientes con disfunción hepática avanzada suelen presentar diversas alteraciones pulmonares. Algunas, como el tabaquismo, son frecuentemente concurrentes, mientras que otras son consecuencia de la hepatopatía. Entre éstas encontramos la hipoxemia por ascitis, la atelectasia compresiva por derrame pleural, la neumonía por aspiración consecuencia de las alteraciones de la consciencia, etc.1.
En los últimos tiempos se ha acuñado el término «síndromes hepatopulmonares», que distingue especialmente 2 que son consecuencia pulmonar de la disfunción hepática, tienen características propias y grave relevancia clínica.
Los pulmones se encuentran «aguas abajo» del hígado. El torrente sanguíneo que fluye del hígado y sistema portal atraviesa el lecho pulmonar, de modo que los cambios en intensidad de flujo o en sustancias vertidas o no metabolizadas por aquéllos pueden afectarlos profundamente2. En efecto, los pacientes con hipertensión portal muestran un estado cardiovascular hiperdinámico, con circulación pulmonar aumentada. Por otra parte, existe un desequilibrio entre sustancias vasodilatadoras y vasoconstrictoras que altera la circulación pulmonar. Estas sustancias, en circunstancias normales, no llegan al pulmón, pero en este caso son producidas o no aclaradas por la circulación esplácnica o el hígado cirrótico en cantidades excesivas3.
En síntesis, en los pacientes con hepatopatía grave es frecuente la existencia de 2 síndromes pulmonares con características propias: el síndrome hepatopulmonar y la hipertensión portopulmonar. Ambos constituyen los extremos de un amplio espectro de vasculopatía pulmonar que va desde la vasodilatación extrema a la vasoconstricción. Si bien guardan relación patogénica con la hipertensión portal, tienen mecanismos fisiopatológicos claramente diferenciados y exactamente opuestos.
EL SÍNDROME HEPATOPULMONAR (SHP)
Este síndrome se caracteriza por una tríada clínica consistente en: a) disfunción hepática; b) hipoxemia, determinada por una presión arterial de oxígeno (PaO2) menor de 70 mmHg o incremento del gradiente alveoloarterial de oxígeno mayor de 20 mmHg (fracción inspiratoria de oxígeno de 0,2), y c) vasodilatación pulmonar4,5.
La incidencia del SHP es muy variable en función de los autores consultados. La razón de esto estriba en los diferentes criterios usados para diagnosticarlo; se proponen valores entre el 4 y el 29%1,6,7. No parece existir una relación clara entre el grado de hipoxemia observado y el grado de disfunción hepática. No obstante, la PaO2 más baja y el mayor riesgo se encuentran en los pacientes en estadio C de Child7. El pronóstico del SHP es malo; la mortalidad se cifra en el 41% a los 2,5 años4.
Fisiopatología
Se han invocado 3 mecanismos diferentes que pueden producir la hipoxemia en el SHP: a) el shunt anatómico verdadero establecido mediante la comunicación de arterias y venas pulmonares evitando el área de intercambio gaseoso; b) el shunt fisiológico, donde la sangre venosa perfunde alveolos no ventilados, y c) la vasodilatación precapilar y capilar del lecho vascular pulmonar que imposibilita la adecuada transferencia de las moléculas de oxígeno al torrente sanguíneo venoso8.
Si bien las alteraciones patológicas que producen el SHP son variadas, el resultado final es una vía común de cambios vasculares que facilitan el cortocircuito de sangre desoxigenada desde la circulación venosa a la arterial. Se considera que el tercer mecanismo la alteración de la difusión del oxígeno es la causa más común de la desaturación. Se ha propuesto también sustituir el término clásico de shunt intrapulmonar por el de desequilibrio alveolocapilar de oxígeno o alteración de la ventilación-perfusión8,9.
El diámetro capilar pulmonar normal es de 8-15 μm. Pues bien, los pacientes con SHP pueden experimentar dilataciones capilares considerables, con diámetros de hasta 500 #mm. En estas circunstancias, las moléculas de oxígeno deben recorrer una larga distancia desde el alveolo hasta ponerse en contacto con la hemoglobina de los glóbulos rojos que pasan por el centro del capilar a una velocidad mayor por la circulación hiperdinámica. En suma, se reduce el tiempo de contacto necesario para que el hematíe se oxigene convenientemente y se produce la hipoxemia. Sin embargo, cuando se ofrece una atmósfera más rica en oxígeno (fracción inspiratoria de oxígeno de 1), mejora considerablemente la oxigenación de los eritrocitos más alejados y aumenta sensiblemente la PaO2. Esto indica la naturaleza fisiológica, no anatómica, del cortocircuito5.
Por último, en el paciente con hepatopatía crónica coexisten con frecuencia otras enfermedades que contribuyen a acrecentar la hipoxemia, como la enfermedad pulmonar obstructiva o restrictiva, la ascitis, el derrame pleural, la desviación de la curva de disociación de la hemoglobina, etc.
Patogenia
Se cree que la vasodilatación pulmonar es la consecuencia del desequilibrio entre la producción y aclaramiento de sustancias vasoactivas circulantes que el hígado insuficiente es incapaz de detraer. Existen numerosas sustancias implicadas en la vasodilatación, como el glucagón, péptido intestinal vasoactivo, péptido relacionado con el gen de la calcitonina, factor natriurético atrial y atriopeptinas, sustancia P, factor de activación plaquetaria, citocinas y óxido nítrico (ON)10.
La hipertensión portal es clave en la génesis de la vasodilatación pulmonar. En efecto, la hipertensión altera la perfusión intestinal e incrementa la translocación bacteriana, lo que estimula la liberación de diversas sustancias vasoactivas. De entre todas las anteriormente citadas, el ON parece desempeñar un papel fundamental. Mediante hallazgos experimentales también se ha involucrado el sistema de la endotelina en la vasodilatación pulmonar. Se postula una hipótesis muy atractiva por la que, en presencia de hipertensión portal, se produce la síntesis hepática de endotelina 1 y la expresión de los receptores endoteliales tipo B, pero no de los tipo A, en los vasos pulmonares. Esta vía conduce al incremento de la producción de ON, mediante la ON sintetasa endotelial y la consiguiente vasodilatación11,12.
En resumen, desde el punto de vista fisiopatológico el SHP tiene unos mecanismos exactamente contrarios a la hipertensión portopulmonar, que ahora revisaremos. Por otra parte, en la circulación pulmonar del paciente con disfunción hepática crónica, la vasodilatación que conduce a hipoxemia arterial es habitual, en tanto que la hipertensión pulmonar es rara.
Diagnóstico
El proceso diagnóstico del SHP debe comenzar documentando la hipoxemia. Para ello se realiza una gasometría arterial con aire ambiente y en supino; confirmada ésta (PaO2 < 70 mmHg), se procede a completar la tríada diagnóstica comprobando que existe vasodilatación pulmonar.
La ecocardiografía de contraste mediante suero salino inyectado intravenosamente, tras agitarlo con vigor, permite diferenciar los shunts derecha-izquierda pulmonares de los cardíacos. En los estos últimos, las burbujas de aire aparecen rápidamente en la aurícula izquierda, en los 3 primeros latidos. Al revés, en el SHP, las burbujas se ven en la aurícula izquierda entre 3 y 6 latidos después de pasar por la derecha.
Constatada la existencia del shunt, es preciso, ahora, cuantificar su magnitud. La prueba más específica es la gammagrafía con microagregados de albúmina marcada con 99Tc. En un paciente normal las partículas isotópicas, al llegar al pulmón, son retenidas por la red capilar dado su mayor diámetro (20-60 frente a 8-15 #m). Por el contrario, en el SHP la vasodilatación pulmonar permite que muchos microagregados traspasen el filtro pulmonar y alcancen otros órganos. La identificación de señales radiactivas sobre el cerebro evidencia el shunt, que se estima mediante la proporción entre la captación cerebral y pulmonar (normal < 5%).
La angiografía pulmonar se reserva para los pacientes en los que se ha demostrado previamente la existencia de vasodilatación pulmonar con ecocardiografía de contraste y han tenido una respuesta pobre al tratamiento de la hipoxemia con oxígeno al 100% (PaO2 < 300 mmHg). En casos seleccionados se podría considerar la embolización de estas comunicaciones arteriovenosas, que constituyen un verdadero shunt anatómico2,4.
Tratamiento
Puesto que la causa del SHP es la vasodilatación pulmonar, el objetivo de cualquier tratamiento debe ser revertirla. Desafortunadamente, con ninguno de los fármacos ensayados se ha obtenido éxito. Una línea de investigación que suscita gran interés teórico consiste en la inhibición terapéutica del ON mediante diversas isoenzimas de la ON sintetasa, aunque todavía no existen resultados consistentes. Otro planteamiento terapéutico diferente sería mejorar la hipertensión portal. Se han administrado fármacos que disminuyen la presión portal, como los bloqueadores beta o los nitritos, aunque con escaso éxito en la mejoría de la hipoxemia. De igual modo, está por demostrar la eficacia clínica de los antibióticos para disminuir la translocación bacteriana. Finalmente, no se han logrado demostrar los beneficios a largo plazo que pueda aportar la colocación de una derivación intrahepática transyugular portosistémica13.
En conclusión, las diferentes opciones terapéuticas no quirúrgicas aplicadas en el SHP han tenido escaso éxito, de modo que el trasplante hepático queda como la única alternativa realmente eficaz en este momento. Sin embargo, la mortalidad del procedimiento todavía es alta (entre el 16 y el 38% el primer año2), especialmente en pacientes que previamente tenían un valor de PaO2 menor de 50 mmHg y/o un shunt mayor del 20%14. Por otra parte, el buen resultado de la intervención no supone un cambio inmediato para la situación pulmonar. Más bien se considera que entonces comienza un proceso de remodelado vascular, más lento y complejo que una sencilla reversión de la vasodilatación. De hecho, la resolución del SHP, entendida como mejoría de la oxigenación, puede requerir hasta 15 meses e incluso las comunicaciones arteriovenosas macroscópicas no llegar a desaparecer15.
Alcanzado este punto, las 2 cuestiones clave que cabe plantearse serían: ¿a qué pacientes trasplantar?, y ¿cuándo hacerlo? Hasta ahora no hay criterios comúnmente aceptados. Un estudio reciente ha comprobado que la PaO2 entre 50 y 60 mmHg antes del trasplante identifica a los pacientes con riesgo de muerte tras la intervención14. La importancia de detectar tempranamente el SHP y estratificar su gravedad es evidente, pues ya hay establecido un sistema basado en el MELD por el cual, cuando la PaO2 de un paciente desciende de 60 mmHg, se considera el trasplante prioritario16,17.
LA HIPERTENSIÓN PORTOPULMONAR (HTPP)
En el extremo contrario del SHP está la HTPP, que resulta de la vasoconstricción pulmonar excesiva y el remodelado vascular y tiene como consecuencia final la insuficiencia del ventrículo derecho. La existencia de la HTPP se contrasta mediante 3 hallazgos hemodinámicos en pacientes con hipertensión portal: a) presión en arteria pulmonar media (PAPM) mayor de 25 mmHg en reposo y de 30 mmHg durante el ejercicio; b) resistencia vascular pulmonar mayor de 120 din·sg·cm5, y c) presión capilar pulmonar menor de 15 mmHg2.
La prevalencia de la HTPP varía mucho en función de los autores, entre el 2 y el 16% según el criterio utilizado18-20. Cuando se emplean los 3 criterios antedichos, la incidencia se cifra en el 4-5%1,21.
Fisiopatología
La ley de Ohm se aplica a la hemodinámica pulmonar mediante la fórmula R = ΔP/Q, donde la resistencia vascular (R) es directamente proporcional al gradiente de presión transpulmonar (P) e inversamente al flujo (Q).
En los estados circulatorios hiperdinámicos hay un aumento considerable del flujo o gasto cardíacos que, sin embargo, no se acompaña del consiguiente incremento de la presión pulmonar. Se debe a la adaptación vascular a esta situación mediante la disminución de la resistencia vascular pulmonar. En el paciente cirrótico, esta capacidad compensatoria parece agotada y la relación P/Q se hace lineal22.
La lesión vascular pulmonar, en la hipertensión portal, conduce a la pérdida de la capacidad reguladora del tono y estructura (remodelado vascular), lo que genera el incremento progresivo de la resistencia vascular que, luego, se traduce en aumento de presión. La hipertensión pulmonar puede llegar a un punto en que decline el gasto cardíaco y fracase el ventrículo derecho.
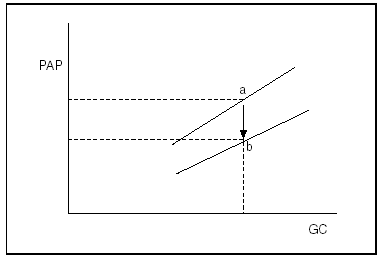
El enfoque hemodinámico adecuado de la HTPP requiere relacionar conjuntamente las principales variables implicadas, como se muestra en la figura 1. En sentido estricto, sólo es posible decir que existe vasoconstricción o vasodilatación pulmonar cuando aumenta o disminuye la presión transpulmonar sin variar el gasto cardíaco. Aplicar este principio resulta fundamental también para determinar el efecto cierto de un tratamiento vasodilatador.

Fig. 1. La resistencia vascular (pendiente) es directamente proporcional a la presión en la arteria pulmonar (PAP) e inversamente al flujo o gasto cardíacos (GC). a: situación basal; b: tras tratamiento vasodilatador.
Patogenia
Todas las formas de hipertensión pulmonar, primaria o secundaria, tienen en común una lesión vascular pulmonar semejante y, por tanto, una clínica y un tratamiento similares. La lesión característica es el remodelado vascular que comienza con la disfunción endotelial, seguida de lesiones del músculo liso, infiltrados inflamatorios, proliferación de la matriz extracelular y coagulación intravascular. El remodelado recuerda, en cierta forma, a la lesión ateromatosa23.
Cronológicamente, la patogenia del proceso comienza con aumento de la reactividad del lecho arterial pulmonar y continúa con el remodelado de la pared vascular. Se postulan distintos factores que podrían desencadenarlo, como: a) el aumento de flujo propio de la circulación hiperdinámica; b) el incremento del volumen vascular pulmonar, y c) el incremento de sustancias vasoconstrictoras en la circulación pulmonar, procedentes del lecho vascular hepatosplácnico24.
Sin embargo, tanto el SHP como la HTPP tienen en común el exceso de producción de endotelina 1, aunque con efectos opuestos, por lo que cabría deducir que la patogenia de cada una de estas 2 complicaciones de la hipertensión portal depende del equilibrio entre la endotelina 1 pulmonar y la producción de ON10.
Por último, se discute si los cambios histopatológicos consistentes en hipertrofia de las capas media e íntima son todavía reversibles pero, cuando hallamos hipertrofia endotelial con cambios plexogénicos, trombóticos y trombóticos, la situación es irreversible2.
Diagnóstico
El diagnóstico definitivo de la HTPP precisa que se confirme el incremento de la presión y resistencia vascular pulmonar. Se trata, pues, de un diagnóstico hemodinámico, por lo que el cateterismo cardíaco derecho es la prueba de referencia. Ahora bien, en la mayoría de los casos la ecocardiografía nos ofrece la primera y más sólida prueba de HTPP. Si la sospecha persiste, el subsiguiente cateterismo ratificará o excluirá terminantemente la HTPP.
La ecocardiografía resulta una prueba excelente, con una seguridad del 95%, y es especialmente útil para excluir la existencia de HTPP con un valor predictivo negativo del 100%25. Permite descartar, en el mismo examen, otra posible causa de hipertensión pulmonar, evalúa la función cardíaca y estima la presión en la arteria pulmonar. La prueba, sin embargo, tiene algunas limitaciones como: a) no es específica y el diagnóstico debe confirmarse con el cateterismo cardíaco; b) hay pacientes sin HTPP en la evaluación previa a la inclusión en lista de espera que la desarrollan mientras esperan el trasplante. De hecho, no se encuentra relación entre la presión pulmonar preoperatoria y perioperatoria en pacientes sin HTPP, y c) no distingue si la hipertensión pulmonar está producida por incremento del tono vascular (HTPP) o éste es normal (estados hipercinéticos)25.
Establecido el diagnóstico, hay que graduar la HTPP para precisar el tratamiento. Si es leve, alcanza una PAPM de entre 25 y 34 mmHg; la moderada, entre 35 y 44 mmHg, y la grave, mayor de 45 mmHg. Hay otras diferencias notables, aunque no siempre consideradas. Así, en la HTPP leve y moderada el gasto cardíaco es normal o elevado y la resistencia vascular pulmonar, moderadamente alta. Por el contrario, en la HTPP grave el gasto cardíaco declina y la resistencia es enorme12.
Tratamiento
No existe tratamiento médico definitivo para la HTPP. En la hipertensión pulmonar primaria, se atribuye al remodelado vascular pulmonar el efecto beneficioso a largo plazo de determinados fármacos como el eprostenol y el ON. Pese a ello, todavía no se ha podido demostrar de modo concluyente que el tratamiento farmacológico mejore la supervivencia de los pacientes con HTPP. Más aún, sorprende el uso infrecuente del epoprostenol en el período pretrasplante, a pesar de ser una indicación más que lógica26. Queda pues el trasplante hepático como el único tratamiento capaz de resolver definitivamente la HTPP, pero ¿qué criterios clínicos y hemodinámicos garantizan el éxito del trasplante?, ¿en que condiciones revierte la HTPP? No lo sabemos a ciencia cierta.
La HTPP significa un riesgo añadido al trasplante; de ahí la necesidad de seguir criterios estrictos de diagnóstico, selección y manejo perioperatorio. Decidida la intervención, se recomienda realizarla en centros de experiencia acreditada en esta complicación12,26.
Los criterios de selección para el trasplante se basan en la evaluación de la presión arterial pulmonar, función ventricular y respuesta a los vasodilatadores. Se considera que la HTPP leve (PAPM entre 25 y 34 mmHg) es reversible tras el trasplante y tiene un riesgo perioperatorio aceptable; por el contrario, cuando la PAPM es mayor de 35 mmHg y la resistencia vascular pulmonar superior a 250 din·sg·cm5, el riesgo es inaceptable1,12,26-28. El dilema surge cuando la PAPM se sitúa entre 30 y 50 mmHg.
El ventrículo derecho es una bomba muy distensible, acomodada a trabajar con flujos altos y presiones bajas, pero es muy sensible a los aumentos de la poscarga que producen hiperpresión pulmonar29. La posibilidad de fallo ventricular es elevada cuando la PAPM supera los 40 mmHg. Conforme a los principios fisiológicos, se considera que 40 mmHg es el valorivel de seguridad para el trasplante hepático27; así que los pacientes que lo superan deben salvar una serie de pruebas antes de ser aceptados.
En primer lugar, se tratan con epoprostenol, en perfusión continua, durante 3-6 meses, tras lo cual se realiza un nuevo cateterismo cardíaco y se evalúa, también, la función ventricular izquierda mediante ecocardiografía de estrés (dobutamina). Si ésta es normal y la PAPM ya es menor de 40 mmHg, se procede a determinar la respuesta del ventrículo derecho a la sobrecarga de volumen: infusión de 1 l de solución salina en 10 min. Si el ventrículo tiene una buena reserva funcional y la PAPM no supera los 40 mmHg, el paciente ya puede ser candidato a trasplante. En caso contrario, se continúa el tratamiento con epoprostenol otros 3-6 meses.
Aun con buena respuesta, es aconsejable continuar el tratamiento mientras se espera el trasplante, pues puede mejorar el remodelado ventricular y vascular durante ese tiempo27. Conviene, en suma, hacer hincapié en que el fin del trasplante es tratar la disfunción hepática, no revertir la HTPP; por lo tanto, mientras aquél se realiza hay que mantener la PAPM dentro de límites razonables con epoprostenol.
Llegados a este punto, antes de efectuar el trasplante es preciso conocer: a) el «grado de plasticidad» de la circulación pulmonar, es decir, cómo responde a los vasodilatadores, y b) la reserva funcional del ventrículo derecho. Importa este asunto porque superar la evaluación preoperatoria no significa salvar el riesgo perioperatorio. Todo lo contrario, los característicos acontecimientos hemodinámicos intraoperatorios resultan especialmente peligrosos en pacientes con HTPP, pues, amén de que surjan cuadros bastante habituales como hemorragia quirúrgica aguda, fibrinólisis, hipercaliemia, acidosis metabólica, etc., que complican el precario equilibrio hemodinámico, los cambios bruscos de volumen pueden resultar fatales30.
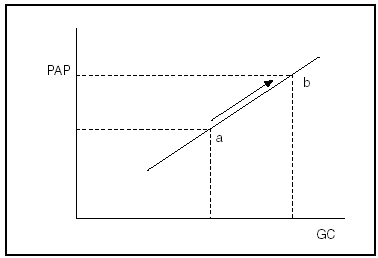
El momento más complicado es la reperfusión del injerto. En pocos minutos las resistencias vasculares sistémicas caen drásticamente, el gasto cardíaco puede duplicarse o más y la presión arterial sistémica disminuir considerablemente30. En el paciente común la función ventricular se mantiene y las resistencias vasculares pulmonares descienden discretamente, por lo que la PAPM sólo aumenta de forma moderada y pronto decae31-34. Por el contrario, en el caso de la HTPP, como se representa en la figura 2, la resistencia vascular, fija y elevada, conduce a que cualquier incremento del gasto cardíaco se acompañe de un aumento similar de la PAPM. Como sabemos, el ventrículo derecho sólo soporta un cierto grado de hiperpresión y puede entrar, súbitamente, en fracaso, con consecuencias fatales.

Fig. 2. Cuando el tono vascular es fijo (misma pendiente), el incremento del gasto cardíaco (GC) se traduce en un aumento similar de la presión en la arteria pulmonar (PAP): punto a hacia b.
Así pues, los objetivos esenciales del mantenimiento intraoperatorio de estos pacientes son: a) conservar una buena función ventricular en todo momento, y b) equilibrar la PAPM. La monitorización hemodinámica ha de ser completa y constante: catéter en arteria pulmonar y ecocardiografía transesofágica; el tratamiento con epoprostenol, igualmente, habitual. La vigilancia y el tratamiento estrictos continuarán en el postoperatorio inmediato; incluso se recomienda continuarlo durante 6 meses, junto con cateterismos cardíacos repetidos27.
CONCLUSIONES
Los pacientes con disfunción hepática avanzada pueden presentar 2 síndromes pulmonares con características propias: el SHP y la HTPP. Ambos constituyen los extremos de un amplio espectro de vasculopatía pulmonar que va desde la vasodilatación extrema a la vasoconstricción. Si bien guardan una estrecha relación patogénica con la hipertensión portal, tienen mecanismos fisiopatológicos exactamente opuestos.
Se considera que la vasodilatación o vasoconstricción es consecuencia del desequilibrio entre la producción o aclaramiento de sustancias vasoactivas circulantes que el hígado insuficiente es incapaz de detraer. De entre las múltiples sustancias investigadas, la hipótesis más atractiva implica a la endotelina 1 y el ON como determinantes esenciales.
El proceso diagnóstico de ambos síndromes está bien definido y consiste en ecocardiografía seguida de gammagrafía en el caso del SHP, y ecocardiografía y cateterismo cardíaco, si procede, en el caso de la HTPP.
El tratamiento del SHP es, únicamente, el trasplante hepático; dado que el pronóstico se ensombrece conforme avanza la hipoxemia, el diagnóstico temprano y el trasplante prioritario resultan fundamentales. El tratamiento de la HTPP con vasodilatadores es sólo paliativo y, también en este caso, el trasplante hepático es la solución definitiva. No obstante, se asumirá el elevado riesgo perioperatorio del procedimiento, exclusivamente, en aquellos casos en que la hipertensión pulmonar es moderada y la vasculopatía responde a los vasodilatadores.

