




INTRODUCCIÓN
El cáncer de páncreas (CP) es, después del cancer colorrectal, una de las neoplasias epiteliales malignas más frecuentes del tracto digestivo y uno de los tumores más agresivos en los seres humanos1,2. Conviene puntualizar, desde el principio, que cuando hablamos del CP nos re-ferimos fundamentalmente al adenocarcinoma ductal (ADC-d), ya que representa más del 95% de los tumores epiteliales malignos de este órgano3. Los ADC acinares, los cisto-ADC y los carcinomas de los islotes de Langerhans apenas suman, entre todos, el 5% restante.
El mejor conocimiento de la biología celular de esta neoplasia devastadora a la que sólo sobrevive un 4% de los pacientes, 5 años después del diagnóstico permitirá plantear, a medio plazo, estrategias diagnósticas y terapéuticas más rentables que las actuales.
Intentar una aproximación al perfil genético y a las bases moleculares de la carcinogénesis pancreática es el propósito de este trabajo.
IDEAS SOBRE CARCINOGÉNESIS
Si tuviésemos que resumir lo que hoy conocemos sobre el mecanismo de realización de la mayoría de los carcinomas podríamos decir que se trata de expansiones celulares autoperpetuables de naturaleza clonal, realizadas en una secuencia multifásica, desde el impacto de numerosos factores etiológicos sobre progenitores divisibles del epitelio diana, a través de la acumulación de varios eventos mutacionales, sufridos por ciertos genes implicados en las funciones celulares vitales4-6. Todo ello, durante una selección competitiva del clono celular dominante, que va trazando un fenotipo progresivamente maligno, tanto en su arquitectura histológica como en su perfil molecular.
Este esquema de realización biológica ha sido probado suficientemente para el cancer colorrectal dada la accesibilidad biópsica en las enfermedades del intestino grueso7, y se sospecha que podría ser también válido para otras neoplasias menos accesibles, como el CP8-10.
La sugerencia de una evolución histogenética multifásica del ADC-d pancreático está apoyada por 3 grupos de argumentos, que desarrollaremos en los apartados siguientes. En primer lugar, la identificación y la estratificación de las posibles lesiones precancerosas pancreáticas; en segundo lugar, la definición de un claro perfil genético-molecular para el ADC-d de este órgano y, en tercer lugar, el hallazgo de que los eventos mutacionales que definen este perfil se han ido adquiriendo y acumulando, con un cierto orden, durante la secuencia evolutiva de las lesiones histológicas progresivamente displásicas.
LESIONES PRECANCEROSAS DEL ADENOCARCINOMA DUCTAL
Se conoce, desde hace tiempo, que algunas enfermedades pancreáticas, en teoría benignas, son situaciones proclives al desarrollo de un CP. Entre ellas destaca la pancreatitis crónica hereditaria originada por mutación del gen que codifica la síntesis del tripsinógeno catiónico, en la medida en que, hasta la edad de 70 años, llegan a desarrollar un CP un 40% de los casos. El riesgo de las pancreatitis crónicas esporádicas es mucho menor, ya que afecta al 4% de los pacientes durante los 20 años siguientes a su diagnóstico3,11.
Algunas raras neoplasias quísticas pancreáticas son procesos auténticamente precancerosos, que se presentan como cistoadenomas mucinosos o mucinoso-papilares, y pueden transformarse lentamente en cisto-ADC3.
Sin embargo, el conocimiento de la probable secuencia multifásica del ADC-d proviene, fundamentalmente, de la identificación y la estratificación de las lesiones morfológicas ductales encontradas en zonas pancreáticas «normales», más o menos cercanas a un CP convencional12,13. Estas lesiones parecen representar precursores histológicos de un ADC-d y, recientemente, un comité internacional de expertos las ha designado con la denominación conflictiva de «neoplasias intraepiteliales pancreaticas» (NIP) y las ha ordenado en 4 estadios: estadios 1-A (hiperplasia plana de epitelio mucinoso sin atipias), 1-B (hiperplasia papilar de dicho epitelio sin atipias), 2 (hiperplasia papilar con displasia de «bajo grado») y 3 (hiperplasia papilar con displasia de «alto grado»), difícil de separar del ADC-d in situ14.
SEÑAS GENÉTICAS DE IDENTIDAD DEL ADENOCARCINOMA DUCTAL
A medida que se ha ido investigando el perfil de las alteraciones genéticas que presentan las células del CP, se ha comprobado que, coincidiendo con la imagen histopatológica de ADC-d invasivo, se puede distinguir una acumulación de varios eventos mutacionales que, hasta cierto punto, es característica de esta neoplasia, como a continuación comentaremos8-10,15.
El protooncogén k-ras es una estructura nucleotídica situada en el cromosoma 12p1.2, que codifica, en condiciones normales, una proteína con actividad en trifosfatasa de guanosina, la cual hidroliza el trifosfato de guanosina (GTP) a difosfato de guanosina (GDP). Esta hidrólisis es la base bioquímica sobre la que asienta la transducción de señales intracitoplasmáticas de origen extracelular, hacia efectores intracelulares de acción nuclear16. Las mutaciones puntuales de uno de los alelos de este gen, que actúa como dominante, están presentes en más del 90% de los CP, lo que conlleva una activación por «aumento de función», con efectos carcinogénicos (oncogén k-ras). El CP es el tumor humano que presenta con mayor frecuencia una mutación de este gen17-21.
En una pequeña proporción de casos, en torno al 10%, se ha encontrado una posible activación de otros protooncogenes (MYB, AKT2, etc.)10.
Hay otras estructuras nucleotídicas, dentro del ADN de las células somáticas, que codifican proteínas que impiden a las células genéticamente lesionadas su tránsito a través del «punto de control» situado en la frontera G1-S del ciclo nuclear y, al mismo tiempo, propician su «apoptosis», modulando la acción del gen bcl-2, regulador de la muerte celular programada. Se trata de la familia de genes supresores de tumor que, comportándose como recesivos, necesitan una alteración genética en forma de mutacion y/o deleción bialélica o epigenética en forma de anormal metilación de su segmento promotor para ser inactivados, con lo que se silencian sus proteínas antineoplásicas22,23.
Cuando se estudia el genoma de las células del CP se encuentra, con gran frecuencia, la inactivación de los genes supresores de tumor p16, p53 y DPC48,10,15,24.
El gen p16, situado en el cromosoma 9p2.1, codifica una proteína que se comporta normalmente como antiproliferativa, cuando se asocia a una cadena de biomoléculas en la que participan la ciclina D, una cinasa dependiente de la ciclina (CDK4), y la proteína RB (retinoblastoma)25,26. Hoy sabemos que en el 95% de los CP hay una disregulación «a la baja» de esta vía metabólica por inactivación genética o epigenética del gen p168-10,15,27,28.
El gen p53, situado en el cromosoma 17p3.1, codifica, en condiciones normales, una proteína que actúa, con carácter antineoplásico, al unirse a una secuencia nucleotídica específica del ADN. De esta manera, cumple con el mecanismo de acción de los genes supresores de tumor, anteriormente comentado, por lo que recibió el título coloquial de «guardián del genoma»29-33. Hoy sabemos que cerca del 75% de los CP presentan algún tipo de inactivación estructural y/o funcional de este gen supresor8-10,15,21,34.
El gen DPC4 (deleted in pancreatic carcinoma, locus 4) es también una estructura nucleotídica del ADN, situada en el cromosoma 18q2.1, que codifica normalmente una proteína que pertenece a las que sintetizan los genes de la familia SMAD, altamente conservados a lo largo de la evolución de los seres vivos. Estas proteínas, y entre ellas la DPC4/SMAD4, se engarzan en una cascada enzimática que regula la actividad antiproliferativa del factor de crecimiento transformante beta (TGF-b)35,36. La inactivación del gen DPC4 se ha encontrado hasta en un 50% de los CP, con lo que se bloquea el avance celular a través del «punto de control» G1-S y se dificulta el sano ejercicio de la «apoptosis» de células genéticamente defectuosas8-10,15.
En una pequeña proporción de CP se ha detectado una inactivación de otros genes supresores de tumor, como el BRCA2 (breast cancer, locus2), en un 7% de los pacientes, y en una proporción menor la de otros genes de esta familia (DCC, STK11 y FHIT)10.
Es bien conocido que la replicación del ADN es un acontecimiento que tiene lugar en la fase S del ciclo nuclear, gracias a la acción enzimática de la ADN-polimerasa, que «empareja» adecuadamente a las bases purínicas y pirimidínicas. De vez en cuando, falla la precisión de esta enzima, lo que crea una inestabilidad genómica que propicia la mutagénesis, si no se repara rápidamente. En los seres humanos, se ha identificado varios genes (MLH1, MSH2, PMS1, MMS2, etc.) cuyos productos proteicos actúan de manera coordinada y secuencial en la reparación del ADN «desparejado»37. El hallazgo de una elevada «inestabilidad de microsatélites» (IMS) es una posibilidad técnica que permite demostrar la existencia de eventos mutacionales en los genes reparadores del ADN «desparejado». Así, un alta IMS se ha demostrado en un 3-13% de los CP. Por el momento, son muy escasos los estudios genéticos más exhaustivos al respecto, aunque se sospecha la pérdida de expresión de la proteína MLH1, en algunos casos9,10,15,38,39. Parece que los CP con errores de replicación del ADN (células RER+) se asocian con una histopatología escasamente diferenciada con un patrón de crecimiento sincitial y una expresión del gen k-ras de «tipo silvestre»40.
Por último, en el tejido de un ADC-d pancreático se suele encontrar una hiperexpresión de algunos factores de crecimiento y otras biomoléculas, cuya síntesis está codificada por genes que no se incluyen dentro de las familias anteriores. Entre ellas, se encuentra el incremento de los factores de crecimiento epidérmico (EGF), hepatocítico (HGF) y transformante alfa (TGF-a), y algunos receptores de membrana (EGF-r, Erb-B2, etc.)15,41.
Dentro de este último grupo de proteínas bioactivas, tiene una especial significación la reactivación funcional de la enzima telomerasa encontrada en el tejido patológico del CP, tal como ocurre en otras neoplasias42. Como es sabido, los telómeros son estructuras genéticas situadas en el extremo de los brazos cromosómicos que se van acortando tras cada mitosis, dada la replicación incompleta del ADN a este nivel. La enzima telomerasa intenta compensar esta pérdida potenciando la síntesis de ADN telomérico. De todas maneras, la activación de esta enzima es casi indetectable en la mayoría de las células somáticas, salvo en algunos sistemas celulares altamente multiplicativos (hemopoyesis, criptas intestinales, etc.). Algo parecido ocurre en los tejidos cancerosos y, entre ellos, en el CP, en los que la reactivación de la telomerasa podría ser crucial para la emergencia de células neoplásicas «inmortales»15.
Desconocemos si todos estos acontecimientos de hiperexpresión de ciertos factores de crecimiento, de sus receptores membranarios o de enzimas facilitadoras de la inmortalización clonal son consecuencia de eventos genéticos, epigenéticos o meros reajustes funcionales fisiopatológicos.
Bien es cierto que el ADC-d pancreático es un tumor que luce una marca genético-molecular muy exclusiva a título de «seña de identidad». Y esto es así, en la medida que: a) más del 90% de los casos acumula en sus células dos eventos, que raras veces se combinan en otras neoplasias, como son la mutación del k-ras y la inactivación del p16; b) más del 80% de los mismos presenta, junto con los eventos precedentes, la supresión funcional del p53, y c) la acumulación de los 3 acontecimientos anteriores, junto con la inactivación del gen DPC4, se puede descubrir en más del 70% de los CP.
CORRELACIÓN HISTOGENÉTICA DE LA CARCINOGÉNESIS PANCREÁTICA
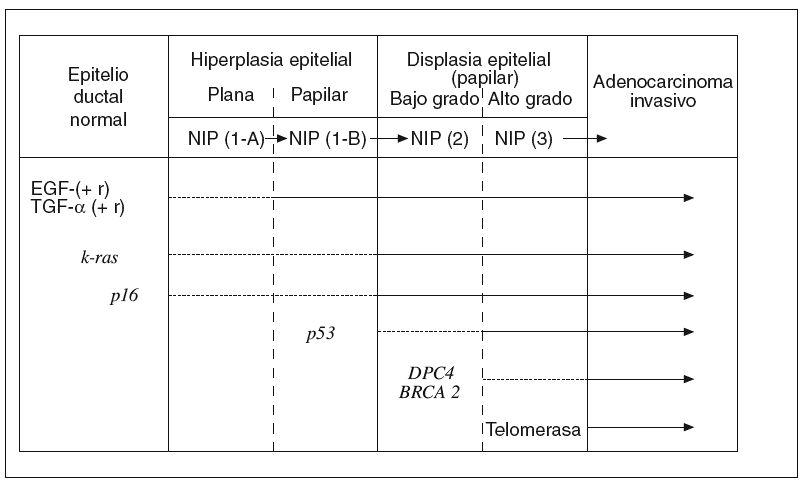
Hay una creencia bastante generalizada de que para la gestación de cualquier cáncer es más importante la «acumulación» de determinadas alteraciones genético-mutacionales que el «orden» en que éstas se producen. Sin embargo, a medida que se ha ido conociendo la secuencia desde el pólipo adenomatoso benigno del colon hasta el carcinoma colorrectal, se ha comprobado que hay un cierto orden en la acumulación de aquellos acontecimientos7,43. Lo mismo empieza a ocurrir respecto a los conocimientos sobre el desarrollo del ADC-d pancreático desde los estadios precancerosos ordenados en la clasificación NIP14. A continuación resumiremos brevemente lo que hoy conocemos sobre la secuencia histogenética de la carcinogénesis pancreática10,15, que hemos tratado de representar en la figura 1.
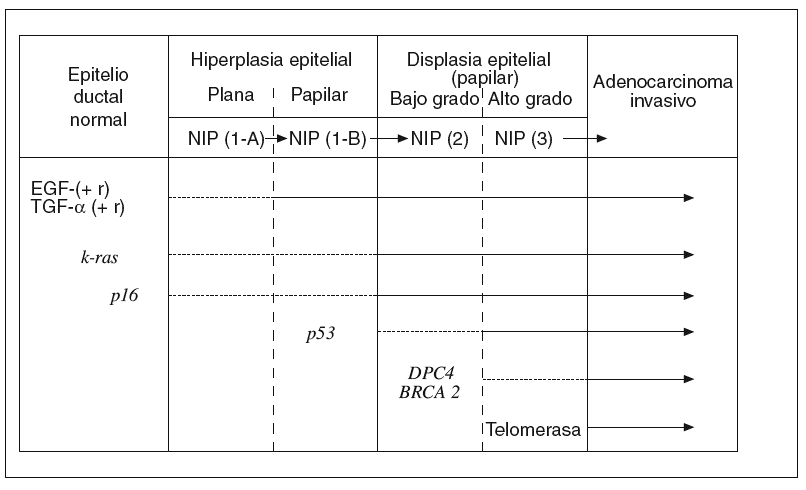
Fig. 1. Carcinogénesis pancreática: secuencia histogenética multifásica. EGF: factores de crecimiento epidérmico; NIP: neoplasias intraepiteliales pancreáticas; TGF-a: Transformante alfa.
El epitelio ductal pancreático, en condiciones normales, es un sistema celular quiescente en el que participa la acción antiproliferativa de naturaleza paracrina ejercida por el TGF-b. Por el contrario, cuando este epitelio sufre el impacto de algunos factores etiológicos que actúan como carcinógenos, se pone en marcha el primer escalón de esta secuencia con una hiperactividad proliferativa del epitelio dirigida por la hiperexpresión de algunos factores de crecimiento (EGF, HGF, TGF-a, etc.), junto con la de ciertos receptores de membrana (EGF-r, Erb-B2, etc.). De esta manera, se cierra un círculo de activación autocrina y paracrina de un epitelio que aún es morfológicamente normal o, a lo sumo, se encuentra en estadio NIP-1A15,41,44.
Junto a estas alteraciones biológicas, empieza a aparecer el primer acontecimiento genético claramente mutacional; concretamente la activación del protooncogén k-ras, que se limita, en un principio, a un número reducido de lesiones en estadio NIP-1A para ir aumentando, durante los estadios siguientes progresivamente displásicos, hasta alcanzar al 90% de los ADC-d invasivos10,15,45-49. La mutación del k-ras, convirtiéndose en un auténtico oncogén, se ha encontrado, en ocasiones, en algunos casos de pancreatitis crónica con hiperplasia ductal sin atipias50.
En los estadios intermedios de la secuencia histogenética es donde comienza a expresarse la inactivación de genes supresores de tumor protagonistas de la carcinogénesis pancreática10,15,51. Así, la inactivación del gen p16 empieza a detectarse en los estadios NIP 1-A o 1-B, la del gen p53 aparece en el estadio NIP-2, y la del gen DPC-4 en el estadio NIP-3. De manera progresiva, el silencio funcional de estos genes alcanza al 95, 75 y 50% de los ADC-d, respectivamente. La inactivación excepcional del gen supresor BRCA2 comienza a plasmarse también en este último estadio de displasia precancerosa de «alto grado»15.
En la fase final de esta secuencia histogenética es cuando suele observarse con claridad la reactivación funcional de la enzima telomerasa, que algo tiene que ver con la inmortalización de las células cancerosas15.
Por último, un acontecimiento biológico, que parece que dirigen las propias células del ADC-d, es la llamada «reacción desmoplásica». Ésta consiste en una activación y una proliferación de los fibroblastos de la estroma peritumoral, con depósito local de componentes de la matriz extracelular, como el colágeno y las metaloproteinasas, al mismo tiempo que se estimula la angiogénesis tumoral, con lo que se facilita la diseminación metastásica15. Se tiene cierta evidencia de que son las propias células del CP las que dirigen esta «reacción desmoplásica», a través de la secreción del TGF-b a los epacios tisulares vecinos. Hay que confesar que, a día de hoy, se desconoce el significado biológico íntimo de este curioso proceso que podría ser la última fase local del «devastador» programa tumorigénico del CP, o una violenta reacción defensiva a cargo del tejido conjuntivo peritumoral.

