







INTRODUCCIÓN
Las glucogenosis son enfermedades hereditarias que se transmiten de manera autosómica recesiva. Los pacientes con este trastorno tienen glucógeno cuya cantidad, calidad o ambas son anormales, ya que las enzimas que intervienen en su síntesis, regulación o degradación están alteradas. Esto impide el mantenimiento de cifras normales de glucemia en ayunas. Se conocen más de 12 tipos de glucogenosis, de los que algunos (I, II, III, IV, VI y IX) cursan con afectación hepática. Los tumores hepáticos son una complicación tardía, sobre todo de los tipos I, III y IV1-3.
Se presenta el caso de una mujer de 31 años, diagnosticada a los 15 meses de edad de glucogenosis tipo III, que a los 30 años de evolución desarrolló un carcinoma hepatocelular (CHC) con trombosis portal. En la bibliografía médica española no hemos encontrado ningún caso de glucogenosis tipo III asociada a CHC.
OBSERVACIÓN CLÍNICA
Mujer de 31 años, sin antecedentes familiares de interés, que ingresó en febrero de 2000 en el Hospital Donostia por dolor en el hipocondrio derecho y ascitis. A los 15 meses de edad (abril de 1970) había sido hospitalizada en la Clínica Infantil La Paz (Madrid) por distensión abdominal y antecedentes de convulsiones a los 20 días, 5 y 7 meses. El embarazo y parto asistencial habían sido normales. Los datos de la exploración fueron: peso de 7,7 kg, estatura de 68 cm, desarrollo psicomotor normal, estado de nutrición regular, irritable y con «cara de muñeca»; abdomen prominente con circulación colateral y hepatomegalia dura, a 5 traveses de dedo del reborde costal.
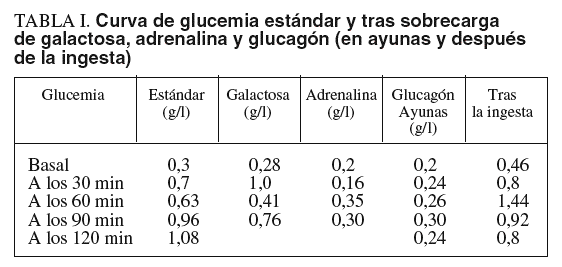
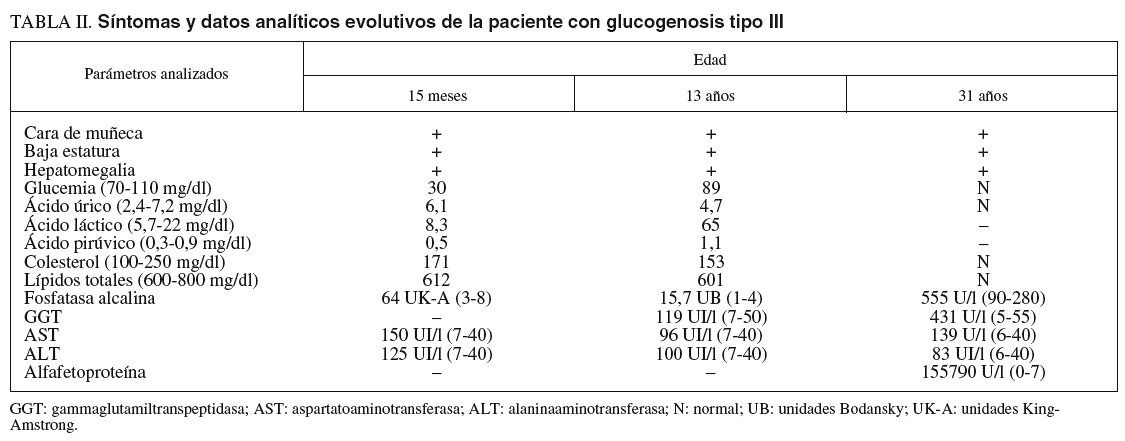
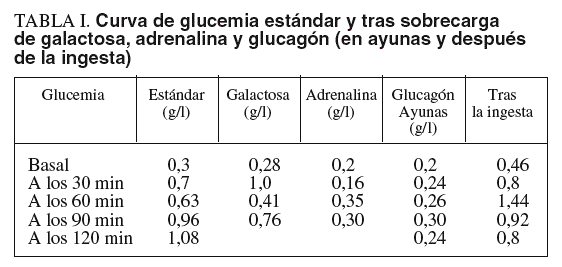
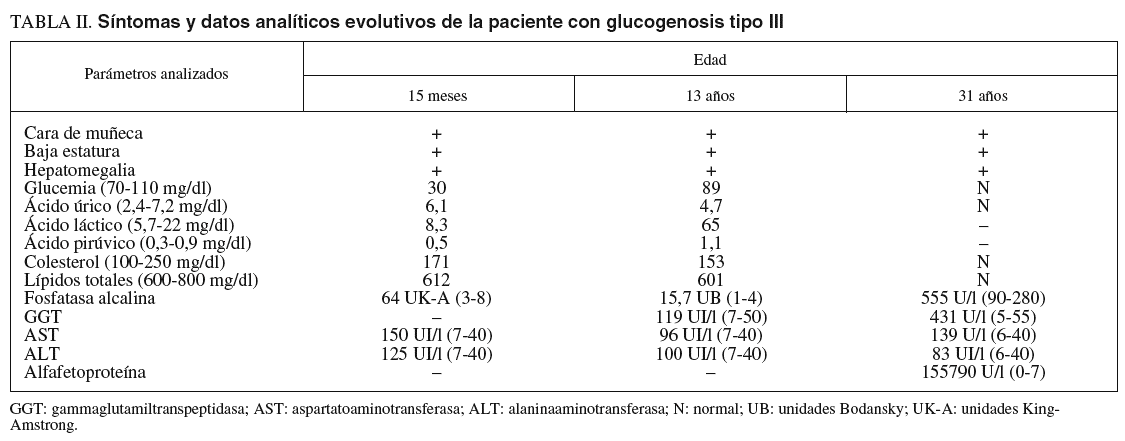
Las pruebas de laboratorio mostraban: hemoglobina de 11,8 g/dl; hematócrito del 35%; leucocitos, plaquetas, coagulación, urea, creatinina, calcio, fósforo, bilirrubina, iones, proteinograma, aclaramiento de creatinina, aminoaciduria global y patrón de aminoácidos, normales. En cuanto al pH y la gasometría, revelaban acidosis metabólica. La glucemia en ayunas era de 0,1-0,3 g/l. Las diferentes curvas de glucemia y otros parámetros bioquímicos evolutivos se recogen en las tablas I y II.
Se efectuó biopsia hepática, en la que se observó una arquitectura alterada por finas bandas conjuntivas, donde se veían canalículos biliares no proliferados. Delimitaban nódulos hepáticos de tamaño regular que carecían de vena centrolobulillar. Los hepatocitos poseían un citoplasma amplio que se teñía intensamente con ácido paraaminosalicílico y ligeramente con Best (figs. 1 y 2). Los hallazgos eran indicativos de glucogenosis. La biopsia muscular reveló fibras musculares estriadas sin alteraciones. No se evidenció depósito de glucógeno en el interior de las fibras musculares.
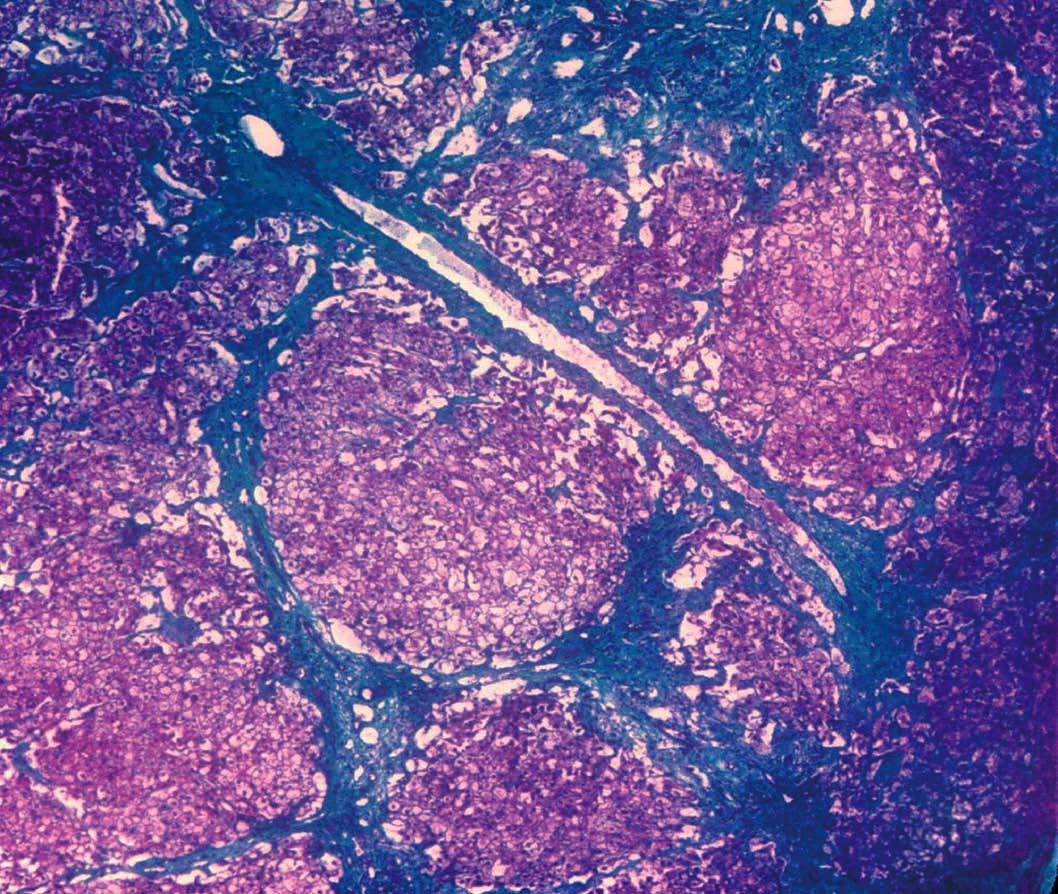
Fig. 1. Biopsia hepática que muestra bandas conjuntivas que delimitan nódulos de tamaño regular que no tienen vena centrolobulillar. (Tricrómico de Masson, *40.)
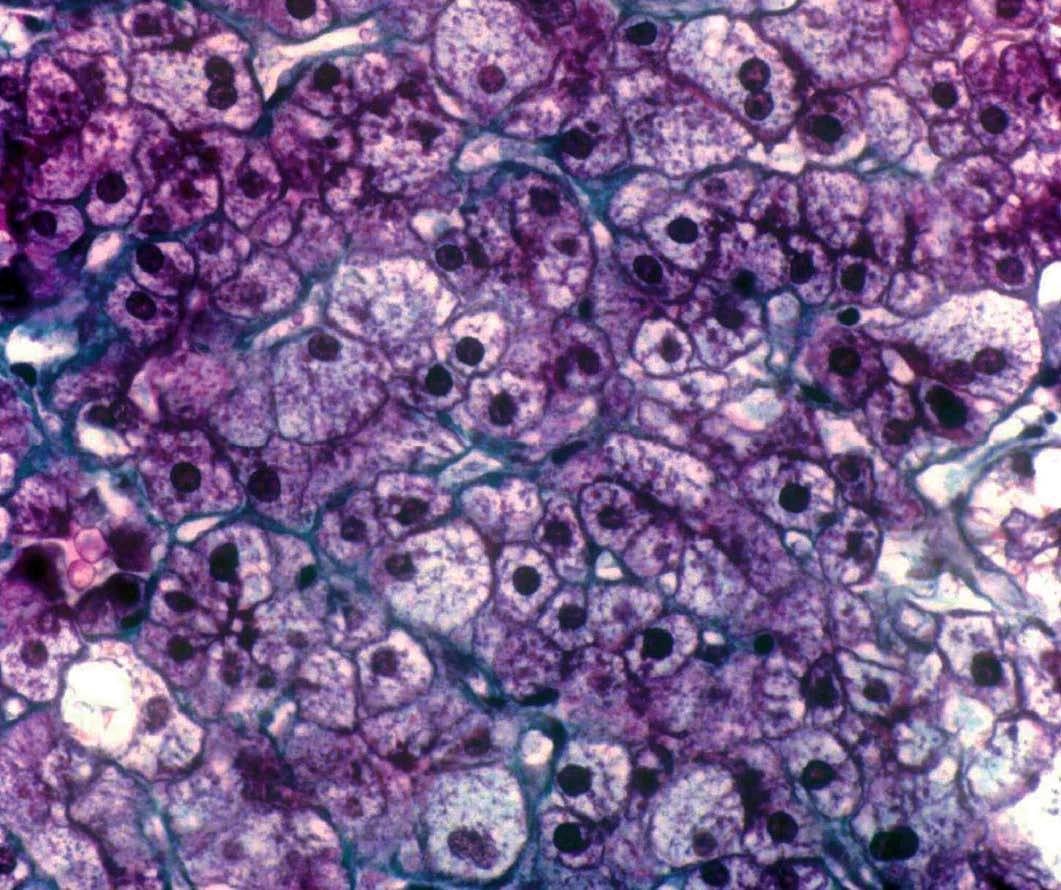
Fig. 2. Biopsia hepática que muestra hepatocitos con abundante citoplasma claro y ricos en glucógeno (aspecto en mosaico, vegetalización). (Tricrómico de Masson, *100.)
A los 10 años la paciente presentó dolor en las pantorrillas, rodillas y los muslos durante 12 meses. La exploración neurológica fue normal, salvo reflejos aquíleos débiles y cutaneoplantares en flexión. El electromiograma y la velocidad de conducción de los nervios ciático poplíteo externo e interno izquierdos fueron normales. Se apreciaba patrón interferencial en deltoides con fuerza muscular disminuida sin actividad espontánea. En los músculos gemelos estos hallazgos fueron menos evidentes. Posteriormente la paciente no volvió a presentar síntomas musculares ni se hicieron estudios electromiográficos.
En varios controles se apreciaron siempre hepatomegalia (5-7 traveses de dedo) y esplenomegalia (7-8 traveses). En las diferentes pruebas de imagen ecografía, tomografía computarizada y resonancia magnética los riñones eran normales y nunca se objetivaron varices esofagogástricas en las sucesivas endoscopias.
A los 31 años la paciente acudió a nuestro servicio por dolor en el epigastrio y hipocondrio derecho, irradiado a la espalda, continuo con períodos de exacerbación, de varias semanas de evolución. En la exploración se apreciaban hepatomegalia dolorosa, dura, nodular (10 cm por debajo del reborde costal), esplenomegalia de 20 cm, ascitis y edema de extremidades con fóvea. Además de los datos analíticos reseñados en la tabla II, destacaron los siguientes: hemoglobina, 13,3 g/dl; hematócrito del 38,8%; leucocitos, 3.820/µl; plaquetas, 80.200/µl; proteínas totales, 6,9 g/l, y gammaglobulina, 1,32 g/l. Los valores de glucosa, coagulación, urea, creatinina, bilirrubina, iones, velocidad de sedimentación globular, inmunoglobulinas, serologías de los virus B y C de la hepatitis, autoanticuerpos y sideremia fueron normales, al igual que la fibrogastroscopia y el tránsito intestinal. La ecografía Doppler abdominal reveló hepatomegalia con ecoestructura no homogénea y ecogenicidad aumentada, sobre todo en el lóbulo izquierdo, hipertensión portal, ascitis, trombosis de la rama izquierda de la porta y estenosis moderada de las venas suprahepáticas. En la resonancia magnética abdominal se apreció hepatomegalia de contorno irregular, así como lesión infiltrante en el lóbulo izquierdo con trombosis portal izquierda, esplenomegalia, ascitis y circulación colateral. La TAC abdominopélvica corroboró los hallazgos anteriores y permitió la realización de una punción dirigida sobre la masa hepática. El informe de la biopsia hepática refería CHC (figs. 3A y 3B). Se indicó tratamiento sintomático y la paciente falleció a los 3 meses.
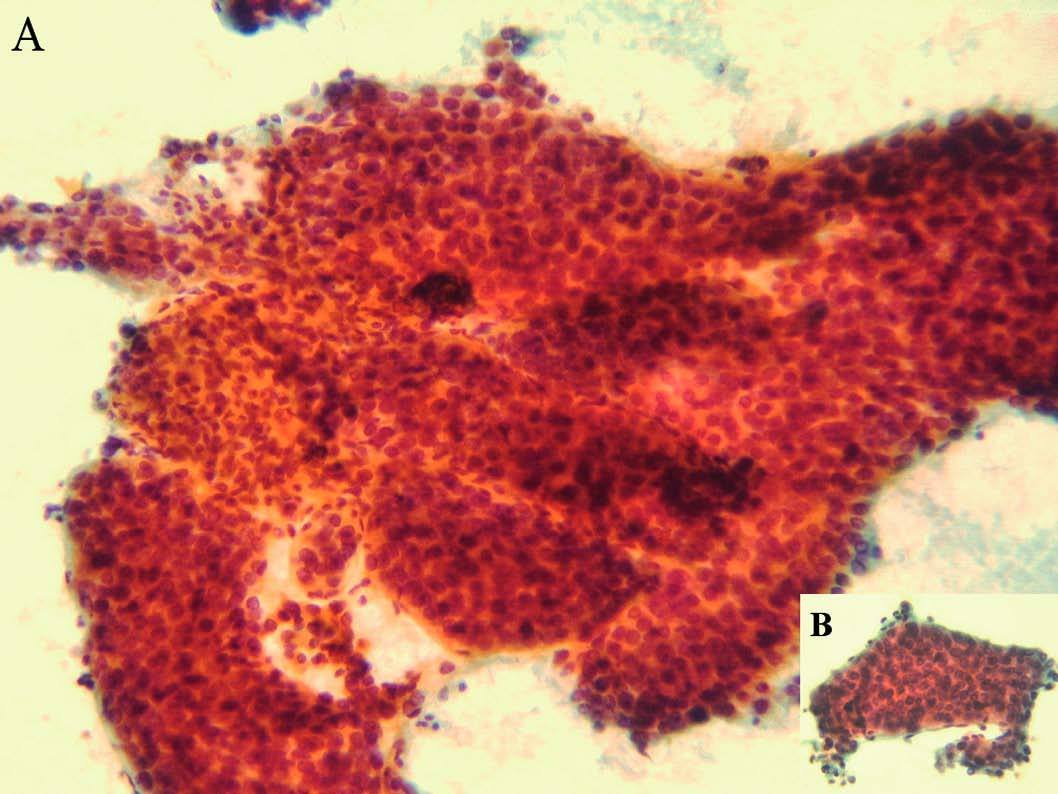
Fig. 3. Hepatocarcinoma bien diferenciado (A): múltiples trabéculas anastomosadas de hepatocitos atípicos. (Papanicolau,*20.) Trabécula de hepatocitos (B) con fenómenos de endotelización periférica. (Papanicolau, *40.)
DISCUSIÓN
La prevalencia de las enfermedades hereditarias por depósito de glucógeno es de un caso por 20.000 o 25.000 nacidos vivos. Los tipos I, II, III y IV son los más frecuentes y corresponden al 90% de todas las glucogenosis.
La glucogenosis tipo III fue descrita por primera vez en 1928 por Snapper y Van Creveld4. En 1956, Illingworth et al5 identificaron el defecto enzimático de la enfermedad. En el tipo III existe una deficiencia de la actividad del complejo enzimático amilo-1,6 glucosidasa y oligo-1,4-1,4 glucanotransferasa, necesario para desramificar el glucógeno después de su degradación por la fosforilasa. Según el déficit selectivo del complejo enzimático, la acumulación de glucógeno se producirá en el hígado, músculo, corazón, nervios o leucocitos. La glucogenosis tipo III, también conocida como enfermedad de Cori o enfermedad de Forbes, se hereda de forma autosómica recesiva. Su incidencia es de un caso por 50.000 o 70.000 personas6 y se asocia a mutaciones del gen AGL (1p21), que codifica el complejo enzimático desramificador del glucógeno. Hay 4 subtipos de glucogenosis tipo III, que dan lugar a diversas manifestaciones clínicas. El 80% de los pacientes pertenecen al subtipo IIIa y tienen afectación hepática y muscular. Un 15% corresponde al subtipo IIIb y sólo tienen afectación del hígado, mientras los subtipos IIIc y IIId son muy raros.
Los niños afectados presentan retraso ponderal y estatural, hipotonía, «cara de muñeca», hipoglucemia tras el ayuno prolongado, cetoacidosis, hiperlipemia y hepatomegalia importante (manifestaciones semejantes a las de la glucogenosis tipo I). La diferencia entre los tipos III y I en niños, en ausencia de afectación renal (más propia del tipo I), radica en la clase de lesión hepática, muscular (miopatía) o cardíaca (miocardiopatía). Tras la pubertad los síntomas tienden a desaparecer, la hepatomegalia se reduce y los pacientes sobreviven hasta la edad adulta. El aumento de transaminasas en la infancia indica lesión hepatocelular (fibrosis e incluso cirrosis micronodular). El descenso en los años siguientes hace pensar que la disfunción hepática se estabiliza y no progresa durante la adolescencia. Se han publicado casos de glucogenosis tipo III asociada a cirrosis micronodular en niños de 8 y 9 años que a los 18 y 22, respectivamente, se encontraban asintomáticos7,8. Desde 1956 hasta el año 2000 sólo se han reseñado 9 casos de pacientes con cirrosis hepática secundaria a glucogenosis tipo III6-15; 4 presentaron hemorragia por varices esofágicas6,10,12,14. La hipotonía muscular, menos evidente en los niños con glucogenosis IIIa, es un síntoma predominante en los adultos que tienen debilidad progresiva de los músculos distales.
Entre las alteraciones detectables en el laboratorio se encuentran el aumento de creatincinasa (si existe afectación muscular), de transaminasas, sobre todo en la infancia, y de lípidos, pero fundamentalmente y desde el principio de la enfermedad, la hipoglucemia. La sobrecarga oral de galactosa o de glucagón origina una hiperglucemia que, en el caso del glucagón, sólo se observa cuando la sobrecarga se ha hecho varias horas después de una comida. Tras el ayuno prolongado la inyección de glucagón no eleva la glucemia. No obstante, las pruebas bioquímicas son sólo indicativas de la enfermedad y el diagnóstico se confirma mediante la determinación de la actividad de la amilo-1,6 glucosidasa y de la oligo-1,4-1,4 glucanotransferasa en el hígado, músculo o fibroblastos. La hipertrigliceridemia, acidosis láctica e hiperuricemia son más propias de la glucogenosis tipo I que de la del tipo III.
La biopsia hepática muestra acumulación importante de glucógeno con puntos de ramificación en los hepatocitos, lo que les confiere un aspecto de células vegetales. En la glucogenosis tipo III se pueden observar fibrosis portal y/o septal, cirrosis y, en menor grado, infiltración grasa. Las técnicas inmunohistoquímicas son útiles para poner de manifiesto el depósito de glucógeno en el hígado y los músculos. En la biopsia hepática o con las técnicas de imagen es infrecuente apreciar adenomas.
La evolución natural de la glucogenosis tipo III se caracteriza por episodios de convulsiones, hipoglucemia y cetoacidosis durante la infancia, y complicaciones tardías (hepáticas) en la edad adulta. La incidencia de adenomas hepáticos en los adolescentes con glucogenosis tipo III es del 25% y siempre en varones1. Sin embargo, en el tipo I son frecuentes (22-75%) y su transformación a CHC ocurre en un 10% de los casos16-18. La cirrosis hepática sintomática es más habitual en los pacientes mayores y puede preceder al CHC. Se conocen 3 casos de CHC sobre cirrosis6,11,15. El primero correspondía a una mujer de 33 años diagnosticada de glucogenosis tipo IIIb a los 2 años y de cirrosis a los 7, a la que se realizó trasplante hepático a los 33 años por cirrosis avanzada. En el hígado explantado se evidenció un CHC6. En el segundo y tercer casos el CHC apareció también en adultos con cirrosis evolucionada11,15. La supervivencia mayor de los pacientes por un tratamiento adecuado en la infancia (dietas ricas en proteínas, alimentación intragástrica nocturna y derivación portocava) y la alteración secundaria de las enzimas implicadas en el metabolismo del glucógeno son hipótesis que pretenden explicar la patogenia del CHC en la glucogenosis tipo III.
En nuestra paciente, el CHC se sospechó por elevación de la alfafetoproteína y se confirmó por punción aspirativa con aguja fina mediante TAC, al igual que en otros casos publicados6,15. Los ultrasonidos ayudan a identificar adenomas hepáticos en todos los tipos de glucogenosis16,19, pero no ofrecen seguridad para detectar CHC sobre cirrosis muy evolucionadas6,15. El CHC es una complicación tardía de algunas glucogenosis hereditarias. Como en los pacientes cirróticos, el seguimiento con métodos de imagen y la determinación periódica en suero de las concentraciones de alfafetoproteína deben llevarse a cabo en los pacienres con glucogenosis para intentar diagnosticar un CHC en una fase susceptible de tratamiento y mejorar así su pronóstico.

