









Introducción: El síndrome de Down (SD) es la aneuploidía más frecuente en el humano, los pacientes con SD tienen un riesgo incrementado mayor de desarrollar leucemias agudas. El pronóstico y los resultados son diferentes a los de la población general, los niños con SD tiene resultados más desfavorables en cuanto a la supervivencia.
Material y métodos: Estudio retrospectivo de una cohorte de pacientes con SD y leucemia linfoblástica aguda (LLA) tratados en el Hospital Infantil de México Federico Gómez, entre enero de 2000 y diciembre de 2010. Se analizaron las características al diagnóstico, complicaciones y supervivencia.
Resultados: Se incluyeron un total de 13 pacientes, la morfología más frecuente fue la L2, el inmunofenotipo más frecuente fue de células precursoras B, se documentó remisión en 12 pacientes, la supervivencia global fue de 60%, el grupo de alto riesgo tuvo una supervivencia de 50% a 24 meses.
Introduction: Down syndrome (DS) is the most common aneuploidy in human. DS patients have an increased risk of developing acute leukemia. Prognosis and results are different from the general population; children with DS have worse outcome in terms of survival.
Material and methods: Retrospective study of a cohort of patients with Down syndrome and acute lymphoblastic leukemia (ALL) treated at the Hospital Infantil de México Federico Gómez between January 2000 and December 2010. We analyzed the diagnostic features, complications and survival.
Results: Thirteen patients were included, the most frequent morphology was L2, and B-cell precursor was the most common immunophenotype, remission was documented in 12 patients. Overall survival was 60%, and the high-risk group had a 50% survival at 24 months.
Introducción
El síndrome de Down (SD) o trisomía 21 constitucional, es la aneuploidía humana más frecuente, con una incidencia de uno en cada 700 nacimientos. Cerca de 80 fenotipos clínicos diferentes han sido identificados en las personas con SD. Este síndrome tiene un espectro amplio de anomalías clínicas siendo las más comunes el deterioro cognitivo, las malformaciones cardiacas y la dismorfia craneofacial, por otra parte se ha notado que existen alteraciones en la hematopoyesis como macrocitosis, cuenta anormal de plaquetas y un incremento en la incidencia de trastorno mieloproliferativo transitorio (TMT), leucemia aguda megacarioblástica y leucemia linfoblástica aguda (LLA)1.
Se ha estimado que entre el 4% y 10% de los niños con SD nacen con TMT, una enfermedad clonal que se caracteriza por megacarioblastos inmaduros en el hígado fetal y sangre periférica, aunque el TMT desaparece espontáneamente en la mayoría de los casos, se considera como un síndrome preleucémico y aproximadamente 20% de los niños diagnosticados con TMT, desarrollará leucemia aguda megacarioblástica dentro los primeros 4 años de vida. La historia natural de la leucemia aguda en los niños con SD, sugiere que la trisomía 21 contribuye funcional y directamente a la transformación maligna de las células hematopoyéticas. Sin embargo, es importante señalar que el SD no es un síndrome de inestabilidad genómica clásico, ya que el riesgo general de desarrollar cáncer, en particular tumores sólidos, es menor en estas personas1,2.
La asociación entre SD y leucemia ha sido bien documentada. Los niños con SD tienen un riesgo incrementado de padecer LLA, comparados con la población general. En estas leucemias predomina el fenotipo de células precursoras B, el fenotipo T suele ser extremadamente bajo. En cuanto a la edad al diagnóstico, no existen diferencias con respecto a la población general. Estudios recientes han revelado que la LLA en niños con SD, es un grupo heterogéneo de neoplasias y que tiene características propias, solo una quinta parte de ellas expresa las características citogenéticas que presentan las LLA en la población general. Existen rearreglos que dan lugar a la expresión de un receptor de citocinas (CRLF2), que se detecta hasta en un 60% de los casos de LLA en pacientes con SD, éstas son asociadas con mutaciones adquiridas en la vía JAK-STAT3-5.
Los niños con SD tienen un marcado incremento en la frecuencia de leucemias mieloides y linfoides. Se estima que el riesgo de SD y LLA es 10 a 20 veces mayor, que el de la población general. La razón de incidencia estandarizada varía de acuerdo a la edad, siendo de 56 para edades entre 0 a 4 años y de 10 para el rango entre 5 a 29 años. Este riesgo es mayor para leucemias mieloides agudas, 3.8 veces más en el rango de los 0 a 4 años con respecto a las linfoides, el riesgo acumulado de leucemias a los 5 años de vida es de 2.1%2-4.
Las características clínicas de presentación de la leucemia en la población con SD son similares, con un pico de incidencia a los 5 años de edad. En lo que respecta al inmunofenotipo, existe un claro predominio de leucemias de precursores de células B, con CD10, CD19 y CD79a positivos. A pesar de esto, existe baja prevalencia de las alteraciones genéticas relacionadas con este inmunofenotipo, como son la presencia de BCR/ABL, TEL/AML1 e hiperdiploidía6.
La anomalía citogenética más común en SD y LLA además de la trisomía 21, es la presencia de un cromosoma X extra, que se puede presentar en la mitad de los pacientes. El papel de la trisomía 21 continúa siendo un misterio, debido a que ésta es la alteración estructural cromosómica somática más frecuente en las LLA en población sin SD, sugiriendo que este cromosoma puede facilitar la leucemogénesis.
El pronóstico en los niños con SD y LLA es menos favorable que el de la población sin SD, en parte debido al incremento de la toxicidad, que se manifiesta por eventos de mucositis, infecciones y muerte durante los periodos intensos de quimioterapia. Esta mayor toxicidad se ha relacionado con aumento de los efectos del metotrexato, debido al exceso de actividad del transportador de folatos, codificado por un gen que se localiza en el cromosoma 21; también se ha observado un marcado efecto inmunosupresor de las antraciclinas. Lo anterior ha llevado a considerar que los ajustes en la intensidad de dosis podrían reducir la toxicidad y ser la premisa inicial para mejorar el pronóstico en este grupo de pacientes, sin embargo esto es un error, pues esta reducción en las dosis no ha permitido igualar la supervivencia. Estudios recientes sugieren que el peor resultado de estos pacientes se debe principalmente a las propiedades genéticas de las células leucémicas, por lo que la reducción excesiva de dosis podría no ser apropiada. En la actualidad se sabe que el mal pronóstico es la baja prevalencia de alteraciones citogenéticas de pronóstico favorable, como son la fusión de genes TEL/AML1 y las trisomías de los cromosomas 4 y 106-8.
Material y métodos
Este estudio incluyó niños con SD y LLA diagnosticados en el Hospital Infantil de México Federico Gómez, durante el periodo comprendido de 1º de enero de 2000 al 31 de diciembre de 2010, todos ellos debían ser menores de 18 años y no haber recibido tratamiento previo.
Para los propósitos de análisis, los pacientes se categorizaron en riesgo estándar y alto, de acuerdo a los criterios del National Cancer Institute. El riesgo estándar incluyó niños con edad al diagnóstico entre uno y 9 años, cuenta de leucocitos menor de 50,000/μL. El grupo de alto riesgo incluyó a pacientes con edad menor de un año y mayor de 9 años, con cuenta de leucocitos mayor de 50,000/μL al momento del diagnóstico.
Todos los pacientes fueron tratados de acuerdo al riesgo, con el protocolo que se maneja en nuestra Institución (Protocolo LLA HIM 2004). La fase de inducción incluye los fármacos vincristina, dexametasona, daunorrubicina, l asparaginasa, etopósido, arabinósido de citosina, que va seguida de consolidación con metotrexato a dosis altas, con rescates de ácido folínico, y mantenimiento de acuerdo al riesgo; se administra quimioterapia intratecal triple.
El análisis de supervivencia fue realizado empleando el método de Kaplan-Meier. La supervivencia global (SG) fue medida de la fecha del diagnóstico, a la fecha de muerte por cualquier causa o fecha de último contacto.
Resultados
Se incluyeron un total de 13 pacientes con el diagnóstico de SD y LLA, en el periodo referido. Ocho (61.5%) fueron del sexo femenino, lo que representa una relación hombre:mujer de 1:1.6.
La edad media al diagnóstico fue de 93.6 meses (DE ± 46.3), con intervalo de edad de 12 a 156 meses. Los principales signos encontrados a la exploración física fueron los del síndrome infiltrativo: adenopatías (61.5%), hepatomegalia (23%) y esplenomegalia (23%) (tabla 1).
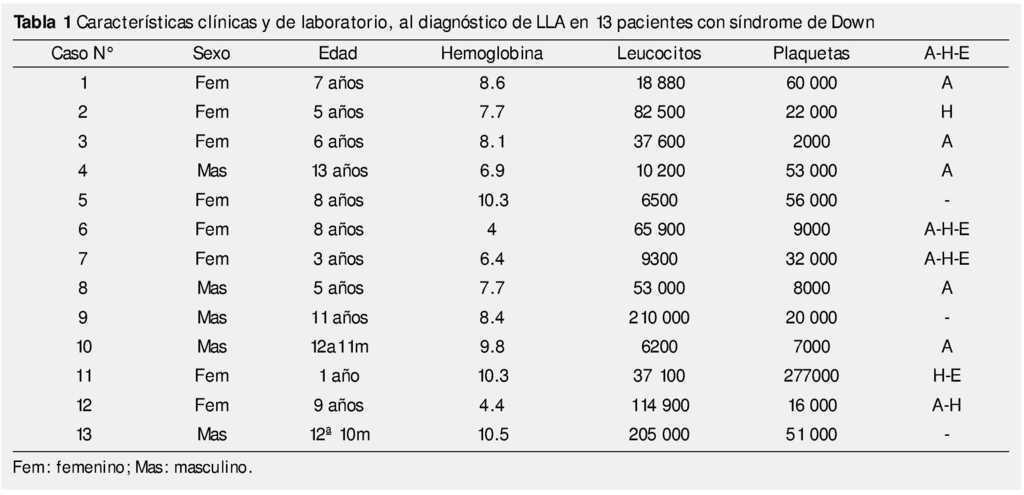
Con respecto a las alteraciones en la biometría hemática inicial, 11 pacientes (84.6%) presentaban anemia, con valores de hemoglobina por debajo de 10 g/dL, cumpliendo en el 18.2% de ellos, con criterios de anemia severa (cifras de hemoglobina menores a 6 g/dL).
Existió trombocitopenia en 12 de los casos, sólo un paciente tuvo una cifra de plaquetas normales. La hiperleucocitosis, definida como una cifra mayor a 100x103/mm3, se observó en 3 casos (23%); 3 pacientes (23%) tenían leucocitos dentro de valores normales para la edad al diagnóstico.
En lo que respecta a alteraciones bioquímicas, todos los pacientes tuvieron cifras de ácido úrico por arriba de las esperadas para la edad y género al momento del diagnóstico, requiriendo manejo para hiperuricemia.
De acuerdo a la clasificación del grupo franco-americanobritánico, 9 casos fueron de morfología L2 (69.2%) y 4 casos L1 (30.8%) (fig. 1).
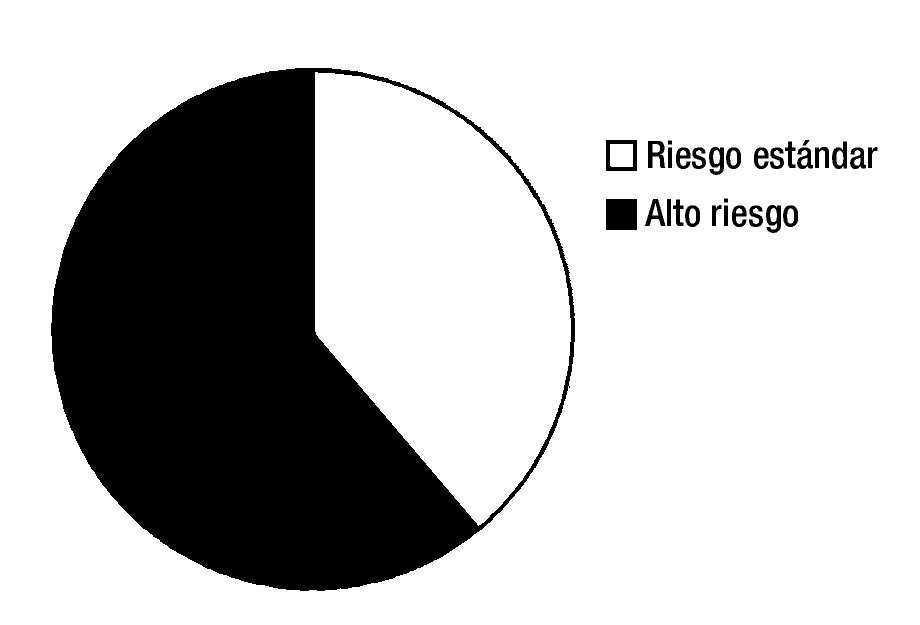
Figura 1. Grupos de riesgo según criterios de Roma NCI. Riesgo estándar y alto.
El inmunofenotipo de estirpe celular B (pre-B), se encontró en 11 pacientes (84.6%) y en 2 casos se cumplieron criterios para clasificar a la leucemia como bifenotípica.
De los 13 pacientes estudiados, 4 tuvieron una alteración citogenética adicional a la trisomía 21, que fue t(1; 19) en 2 casos y t(12; 21), t(9; 22) e hiperdiploidía en un caso para cada una (tabla 2).
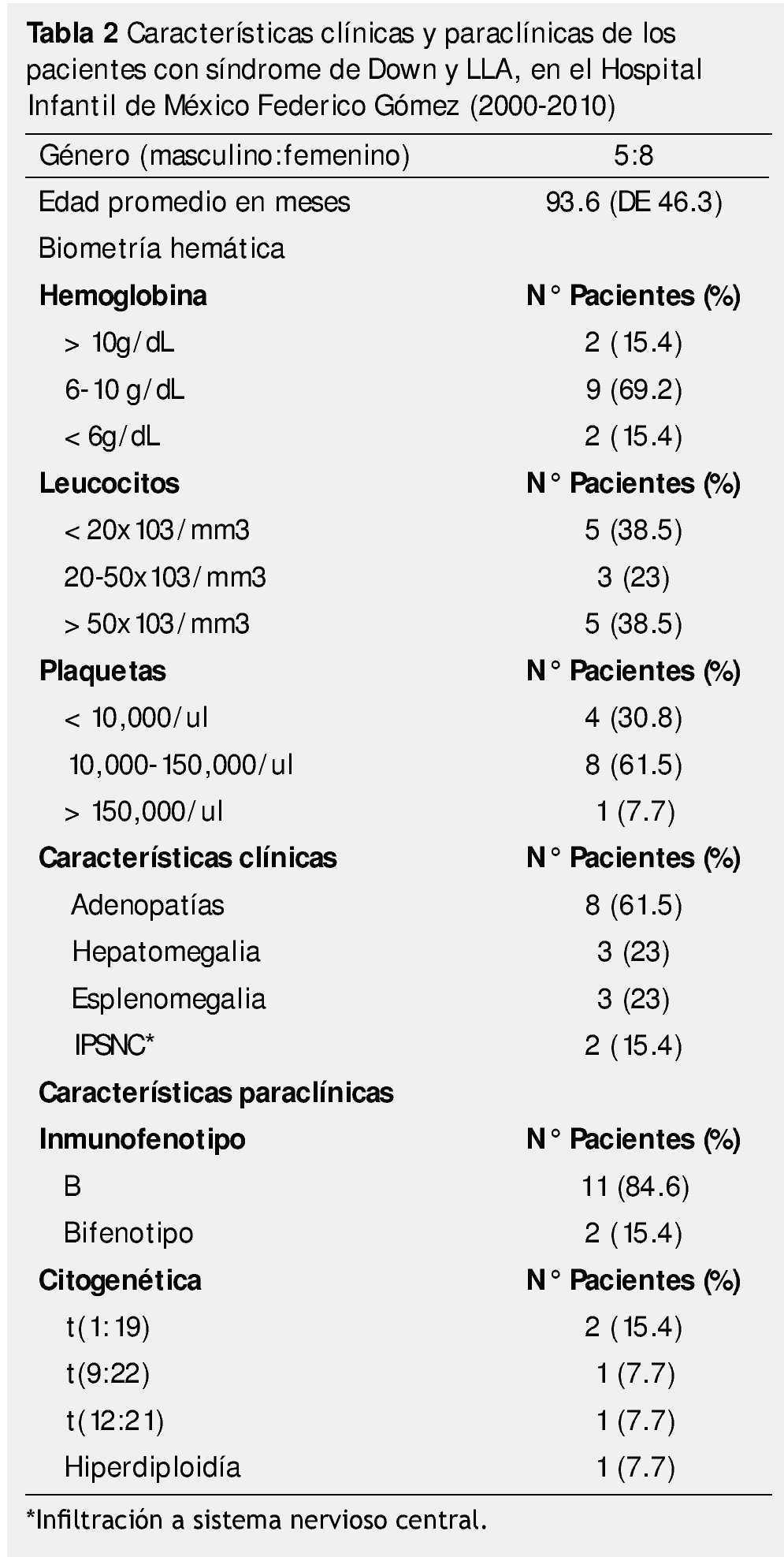
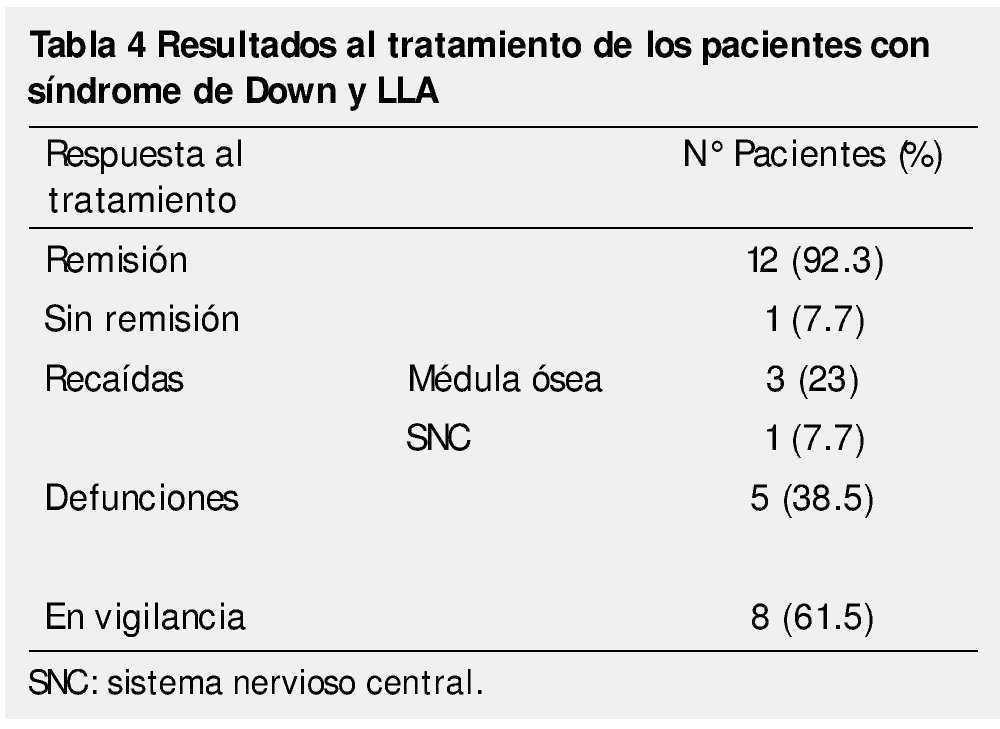
En lo que respecta a la respuesta al tratamiento, 12 niños presentaron remisión morfológica al día 14, evaluada mediante el porcentaje de blastos en médula ósea (< 5%). Sólo un caso mostró resistencia primaria con falla a la inducción y comportamiento refractario, este paciente falleció por complicaciones relacionadas a la progresión de la leucemia.
Cuatro pacientes presentaron recaídas, que fueron a médula ósea en 3 casos y en uno, la recaída fue aislada a sistema nervioso central. Todas ellas ocurrieron durante el primer año de tratamiento, y no remitieron con los esquemas empleados para el manejo de recaídas en nuestra Institución.
Todos los pacientes tuvieron complicaciones relacionadas con el tratamiento. La mucositis y la fiebre asociada a neutropenia fueron las más frecuentes, 12 pacientes (92.3%) presentaron al menos uno de estos eventos adversos, de grados 3 o 4 a lo largo del tratamiento.
Los cuadros infecciosos más frecuentes fueron neumonía, que se presentó en 4 casos, (30.7%), y sepsis en 3 casos (23%). Las complicaciones abdominales más comunes incluyeron colitis neutropénica, que ocurrió en 3 pacientes, y pancreatitis en 2 casos (15.4%). La infección pulmonar por Aspergillus se documentó en 2 de los pacientes (15.4%). Observamos también diabetes mellitus secundaria al uso de esteroide en 2 casos (tabla 3).

Debido a esta toxicidad, algunos pacientes precisaron ajuste en la dosis de quimioterapia o retraso de la misma. Las reducciones de dosis se hicieron únicamente en metotrexato y las dosis de otros fármacos fueron las mismas, que las utilizadas para pacientes sin SD.
A pesar de la buena respuesta temprana al tratamiento, las complicaciones por la inmunosupresión, propia de la enfermedad y adquirida por el tratamiento, generaron una mortalidad del 38.5% en los pacientes incluidos. El choque séptico fue causa directa de muerte en 4 de los 5 fallecimientos de esta serie. El paciente que falleció con progresión de la enfermedad, desarrolló tuberculosis meníngea y neumonía nosocomial, las cuales fueron las causas de muerte registradas (tabla 4).
La SG en el grupo de bajo riesgo fue de 80% a 5 años, y de 55% para el grupo de alto riesgo (figs. 2 y 3).
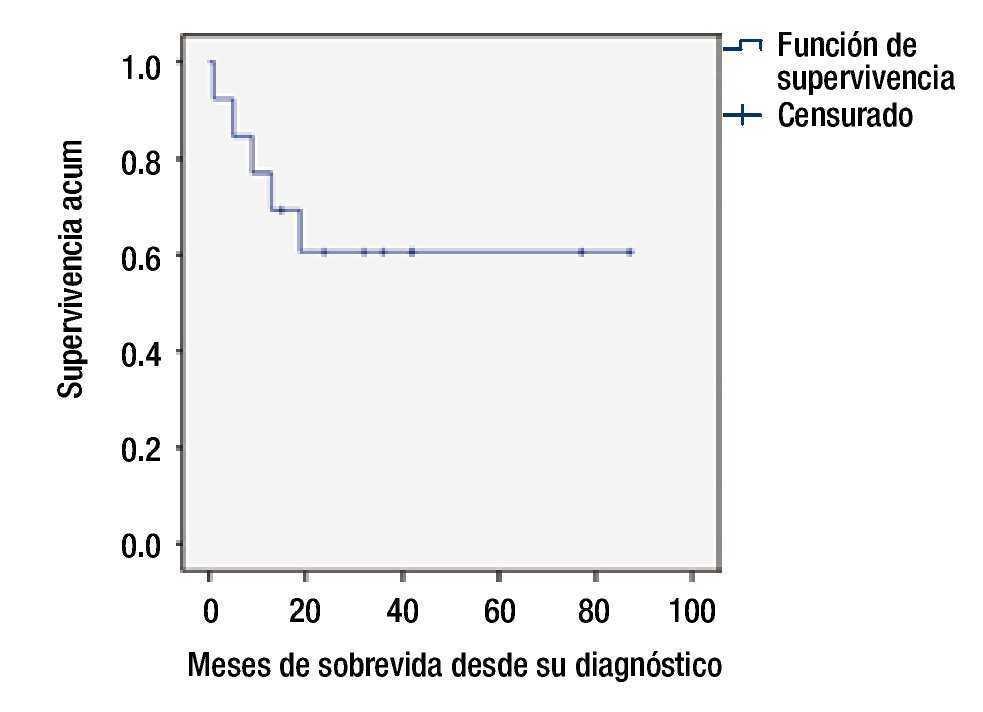
Figura 2. Supervivencia global.
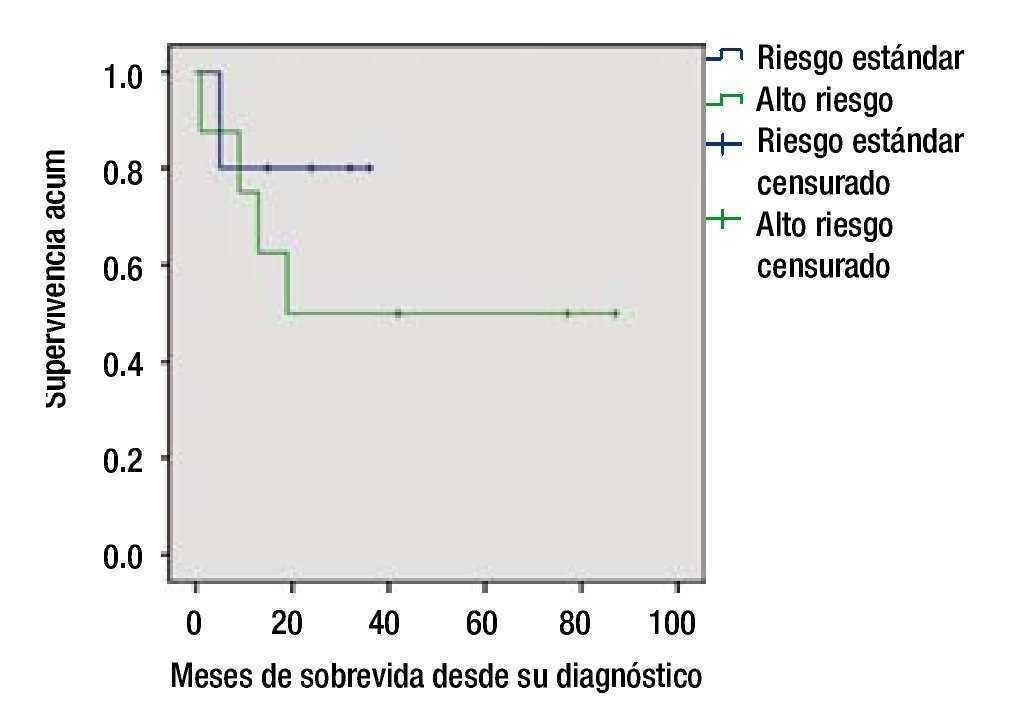
Figura 3. Supervivencia global por grupo de riesgo.
Discusión
Como se ha referido en la literatura médica, el niño con SD y LLA tiene características clínicas y biológicas diferentes, a las de los pacientes sin SD. Una diferencia observada en nuestra cohorte de pacientes fue la edad de presentación, ya que la edad media al diagnóstico fue de 7.8 años, que es significativamente mayor a la de los pacientes con LLA sin SD. El resto de características clínicas fueron similares a las de la los pacientes sin SD, predominando las relacionadas con el síndrome infiltrativo, es de notar que la fiebre no es una manifestación común en este grupo. Las alteraciones en la biometría hemática, que reflejan el grado de disfunción de la médula no exhibieron características distintas a lo reportado en la literatura médica. El inmunofenotipo más frecuente, como se describe en la mayoría de los reportes fue de precursores de células B, en la mayoría de los casos. Encontramos alteraciones cromosómicas adicionales a la trisomía 21 en 4 pacientes; en un niño la presencia de cromosoma Philadelphia.
En cuanto a la respuesta al tratamiento, en este pequeño grupo hubo un paciente en quien nunca remitió la leucemia. En la literatura médica se reporta que con los esquemas actuales de tratamiento, menos del 3% de los pacientes muestran este comportamiento.
Todas las recaídas se presentaron durante el primer año del tratamiento, y en todos los casos desarrollaron un comportamiento refractario. Es muy probable que en este patrón de falla a tratamiento estén implicados factores de resistencia genética.
En la actualidad, sabemos que uno de los factores que influyen en el peor pronóstico de los niños con SD y LLA, es la mayor incidencia de procesos infecciosos a los que son susceptibles debido a las alteraciones del sistema inmune, sumadas a los efectos de la quimioterapia, principalmente los relacionados con los antimetabolitos como el metotrexato y la citarabina, así como con las antraciclinas. Asimismo, los ajustes de dosis que en general se realizan con el objetivo de disminuir los eventos adversos del tratamiento impactan también en el resultado a largo plazo, ya que pueden favorecer recaídas, debido a la disminución de la intensidad de dosis. Las complicaciones relacionadas con el daño a mucosas, neutropenia y fiebre, se presentaron casi en la totalidad de los pacientes de nuestra serie y determinaron la necesidad de reducir las dosis altas de metotrexato, siendo posible que tanto los atrasos en el tratamiento debidos a las complicaciones, como la reducción en la dosis de metotrexato hayan contribuido en el desarrollo de recaídas.
Otro aspecto a destacar es que el niño con SD tiene hiperuricemia como parte de las características biológicas que lo distinguen, y durante el tratamiento del síndrome de lisis tumoral, al diagnóstico o en recaídas, se debe tener en cuenta que no en todos estos pacientes se normalizarán las cifras de ácido úrico a pesar del manejo enérgico del síndrome de lisis tumoral.
La SG en este grupo fue inferior con respecto a la descrita por el Childrens Cancer Group, el cual reporta 75%, pero dentro de un rango aceptable de acuerdo a los protocolos de tratamiento del grupo BFM 81, 83, 86, y 90, donde el resultado fue peor para los pacientes con SD y LLA en comparación con aquellos sin SD.
Conclusiones
El tratamiento de los niños con SD y LLA continúa siendo variable de acuerdo a los protocolos y grupos de tratamiento, se ha observado que tienen características biológicas únicas y que el impacto en el pronóstico no se debe a las modificaciones en la intensidad de dosis, es muy probable que el mayor impacto para igualar los resultados en este grupo sean realizados en un diagnóstico precoz; también es importante mejorar las estrategias del tratamiento de las complicaciones, así como conocer los riesgos a los cuales está expuesta esta población.
Financiamiento
No se recibió ningún patrocinio para llevar a cabo este artículo.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
* Autor para correspondencia:
Hospital Infantil de México Federico Gómez.
Dr. Márquez N° 162, Colonia Doctores, Delegación Cuauhtémoc, C.P. 06720, México D.F., México.
Correo electrónico: phalomi@hotmail.com (Miguel Ángel Palomo-Colli).