





El cáncer de pulmón de células no pequeñas (CPCNP) representa aproximadamente el 75%, de todos los tumores pulmonares. Los genes comúnmente mutados en esta neoplasia son: el EGFR, el KRAS y recientemente, se reportó la proteína de fusión EML4-ALK. Las mutaciones en el EGFR y la fusión proteína de fusión EML-ALK, confieren sensibilidad a los inhibidores de cinasa de tirosina (TKIs), mientras las mutaciones de KRAS confieren resistencia a los estos. La frecuencia de este tipo de mutaciones, varía en diferentes grupos étnicos. Por este motivo, en la actualidad surge el interés de llevar a cabo la genotipificación de cada uno los pacientes con CPCNP, con la finalidad de personalizar el tratamiento de acuerdo a su fondo genético. Por lo cual en este artículo, nos enfocamos a describir mecanismos moleculares presentes en los pacientes con cáncer de pulmón, los cuales, impactarán en la toma de decisión del tratamiento adecuado para el paciente oncológico.
Non-small cell Lung cancer (NSCLC) represents approximately 75% of all lung tumors. Commonly mutated genes in this neoplasia are: EGFR, KRAS and recently was reported the fusion protein EML4-ALK. The mutations in the EGFR and the EML4-ALK fusion protein confer sensitivity to tyrosine kinase inhibitors (TKIs), while KRAS mutations confer resistance to these. The frequency of these mutations varies in different ethnic groups. Is for this reason, it is important the genotyping of the patients with NSCLC, in order to customize the treatment to each patient according to their genetic background, this generate an increase in overall survival. So in this chapter, we describe the molecular mechanisms present in patients with lung cancer, this impact to determine appropriate lung cancer patient treatment.
La carcinogénesis pulmonar es un proceso crónico que involucra múltiples alteraciones genéticas, celulares y tisulares, resultado de modificaciones en genes que regulan el crecimiento, la diferenciación y la apoptosis, lo que finalmente lleva al desarrollo de cáncer invasivo o metastásico. Esta neoplasia es la primera causa de muerte por cáncer a nivel mundial, tanto en hombres como en mujeres.1 En el 2009, se registraron 200 000 nuevos casos de cáncer de pulmón. A nivel mundial, en el 2010 se esperan 1 300 000 casos diagnosticados. De los pacientes con cáncer pulmonar, sólo el 16% sobreviven a cinco años, por lo cual representa un problema grave de salud.2
La principal causa del cáncer de pulmón es el tabaquismo. Sin embargo, existen otros factores de riesgo como lo son: factores genéticos, hormonales, la exposición a metales pesados y al humo de leña.3
El cáncer de pulmón se divide en dos grupos: el cáncer de pulmón de células pequeñas y el cáncer de pulmón de células no pequeñas (CPCNP), este último es el subtipo más frecuente en la población, ya que representa aproximadamente el 75% de todos los tumores pulmonares. El CPCNP se divide principalmente en tres tipos histológicos: el carcinoma escamoso, el adenocarcinoma y el carcinoma de células grandes.1 Esta neoplasia tiene un pronóstico pobre de sobrevida, por la baja efectividad de los tratamientos asociada al desarrollo de resistencia tumoral intrínseca y adquirida, que se manifiesta clínicamente por progresión temprana y respuestas transitorias.
¿ MARCADORES MOLECULARES EN CÁNCER DE PULMÓN
Durante el desarrollo del CPCNP, ocurren diferentes eventos moleculares que incluyen la pérdida de heterocigocidad, cambios epigenéticos, mutaciones en p53, KRAS y en el receptor del factor de crecimiento epidérmico (EGFR), la amplificación del cMET y la inestabilidad de microsatélites, solo por mencionar algunos.3 Es importante destacar, que las mutaciones presentes en el EGFR son prevalentes en adenocarcinomas de no fumadores, mientras que en adenocarcinomas de fumadores lo son las mutaciones en KRAS.4 Estas diferencias son potencialmente importantes, para asignar el tratamiento adecuado al paciente basado en agentes biológicos.
Diferentes estudios moleculares han reportado modificaciones en vías de señalización, que contribuyen a la tumorigénesis del pulmón. Algunas de ellas, involucran a las proteínas EGFR, KRAS, cMET y AKT. El EGFR se ha asociado con la carcinogénesis y progresión del tumor, a través de diferentes mecanismos como la sobre expresión del receptor y el ligando, así como también a través de diferentes mutaciones, las cuales se asocian con la activación de distintas vías de señalización.
¿ VÍA DE SEÑALIZACIÓN DEL EGFR
El gen EGFR se encuentra localizado en el brazo corto del cromosoma 7 y codifica para una proteína transmembranal, con un tamaño molecular aproximado de 170 kDa. Pertenece a una familia de cuatro receptores de membrana, con actividad de cinasas de tirosina (TK): ErbB1 (EGFR, HER1), ErbB2 (HER2/neu), ErbB3 (HER3) and ErbB4 (HER4). Todos los miembros de esta familia, presentan una estructura similar que consiste de tres regiones: una región extracelular, donde se localiza el dominio de unión al ligando, una región transmembrana, donde se ancla a la membrana plasmática y una región intracelular, donde se encuentra el dominio de TK5 (Figura 1).
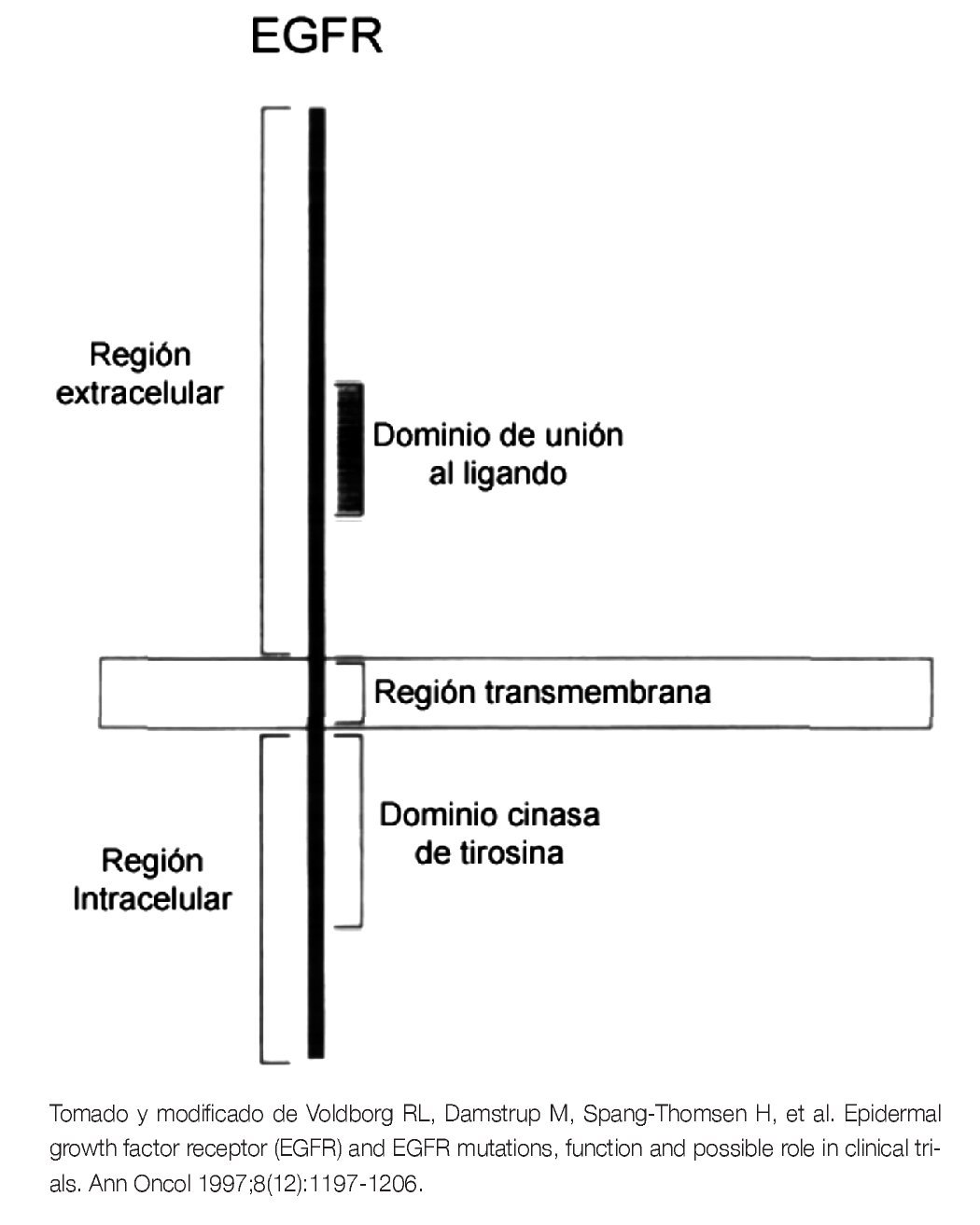
Figura 1. Estructura esquemática del EGFR.
En respuesta a la unión de diferentes ligandos como el factor de crecimiento epidérmico (EGF), el factor de crecimiento transformante alfa (TGF-α), beta-celulina, epiregulina y amfiregulina, el EGFR es capaz de dimerizar con otro receptor del mismo tipo (homodimerización) o bien, con otros receptores de la misma familia (heterodimerización) para inducir la activación de su dominio de TK y llevar a cabo su autofosforilación en cinco residuos de tirosina (Tyr 1173, 1148, 1086, 1068 y 992).6,7 La autofosforilación permite la activación de múltiples vías de señalización rio abajo, como la vía de RAS-RAF-MAPK, la PI3K y la vía STAT, que inducen la regulación de la proliferación e invasión celular, angiogénesis, inhibición de la apoptosis y metástasis (Figura 2).8
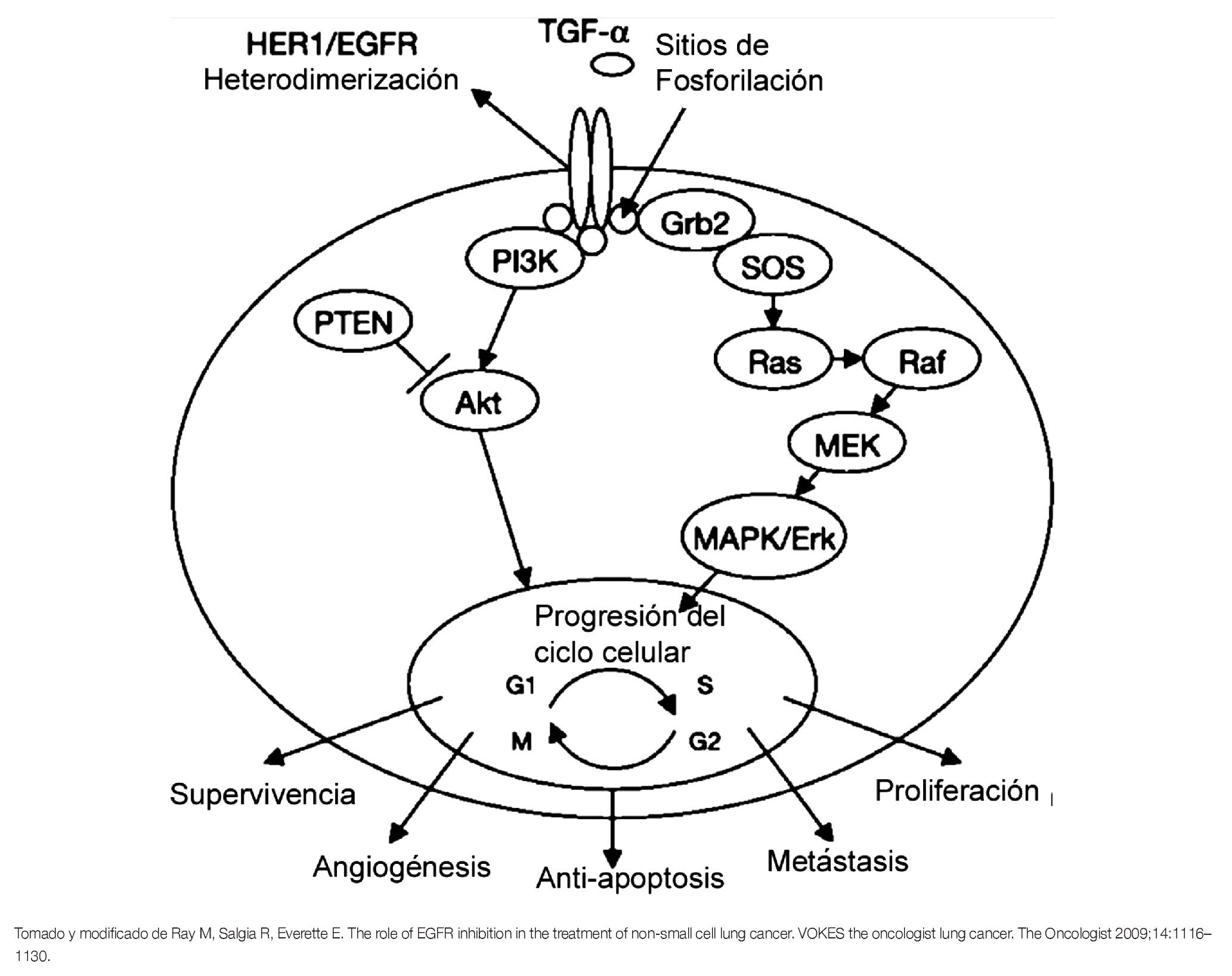
Figura 2. Representación esquemática de la vía de señalización del EGFR.
La vía de RAS-RAF-MAPK activada constitutivamente por el EGFR, influencia la adquisición de un fenotipo maligno a través de la síntesis de DNA y la proliferación celular descontrolada.5 Por otra parte, se han reportado mutaciones en KRAS que aparecen en eventos tempranos del CPCNP, principalmente en adenocarcinomas. Estas mutaciones son mutuamente excluyentes, de las mutaciones del EGFR y están asociadas a la resistencia de los inhibidores de TK del EGFR (EGFR-TKIs).3
La actividad constitutiva del EGFR ha sido observada en más del 60% de pacientes con CPCNP,9,10 y se debe a diferentes mutaciones presentes en el receptor. Estas representan el 50% en no fumadores, con respecto al 10% de fumadores y al 40% en adenocarcinomas, con respecto al 3% de otras histologías.11 Más del 90% de estas mutaciones, están localizadas en los exones 19 y 21 (deleciones y la mutación puntual L858R respectivamente) del EGFR, donde se localiza el sitio de unión al ATP del dominio TK12 (Figuras 1 y 2). Tanto las deleciones del exón 19, como la mutación L858R resultan ser mutaciones de respuesta a EGFR-TKIs, como el gefinitib y erlotinib.11 Debido a la mutación L858R, el gefitinib se une 20 veces más a la mutante L858R, que al EGFR silvestre.
¿ TRATAMIENTO DEL CPCNP
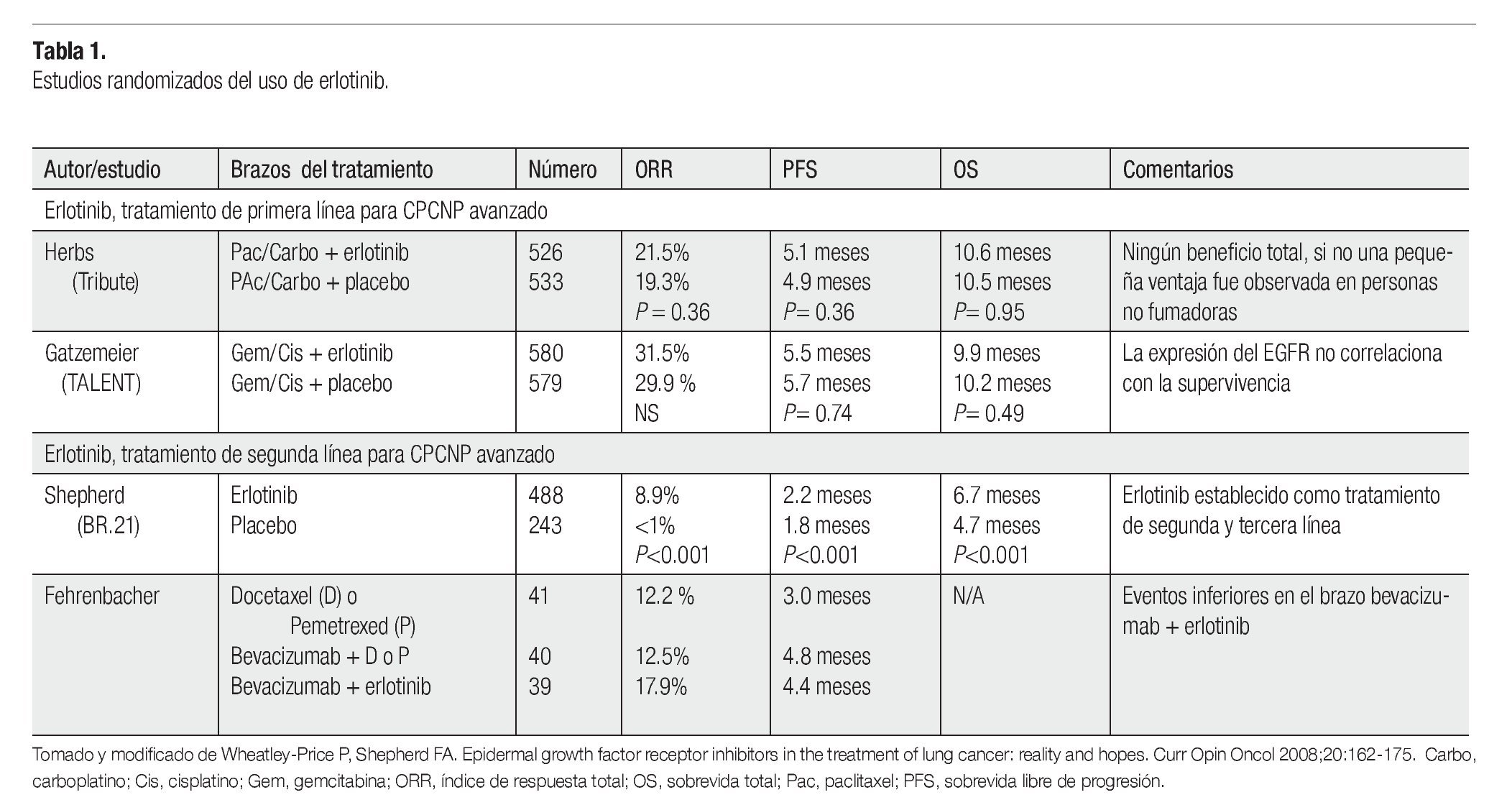
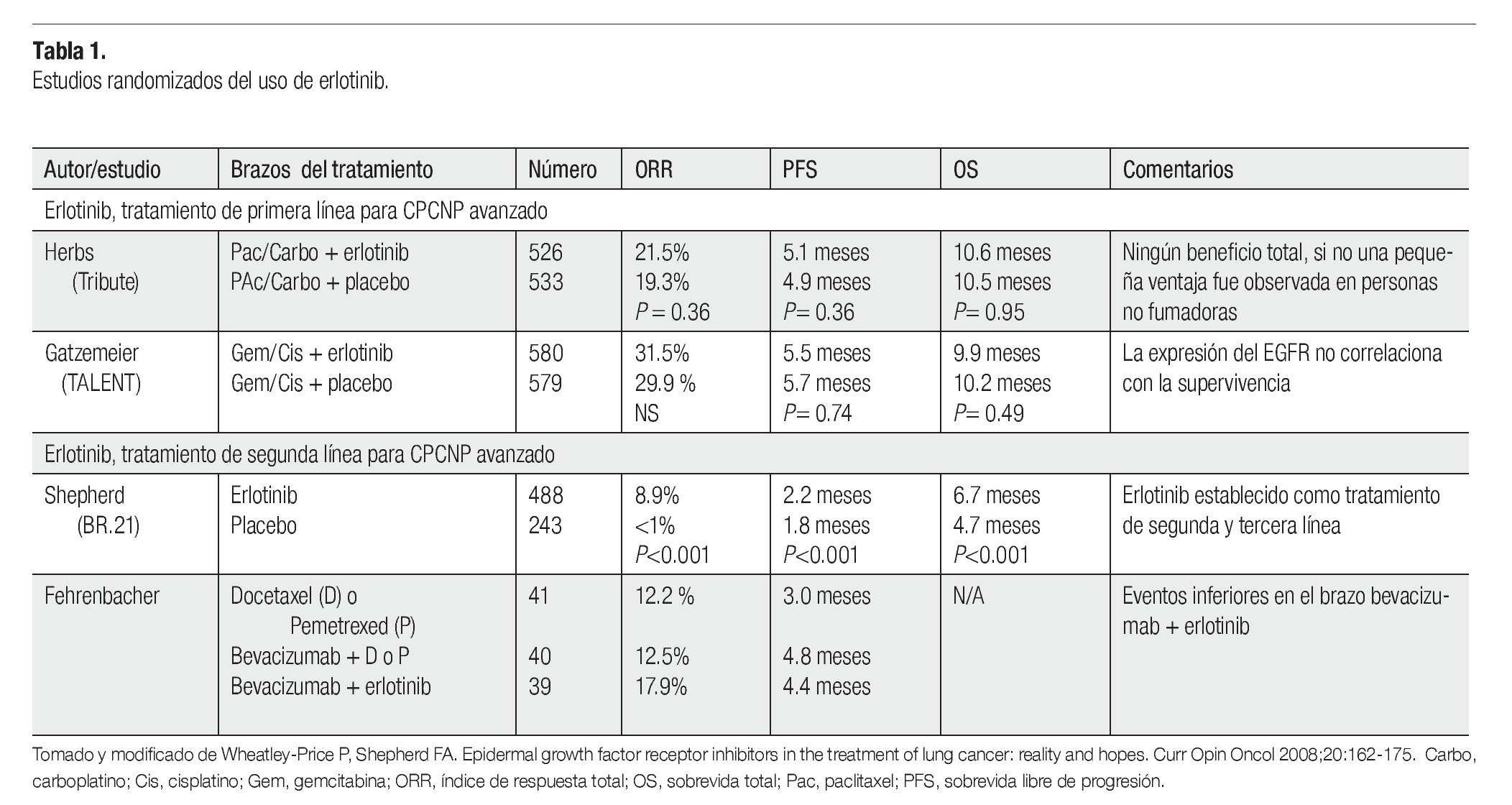
Existen diferentes opciones para el tratamiento del CPCNP, tales como la radioterapia, cirugía y quimioterapia, dependiendo del estadio del tumor. La combinación del cisplatino con agentes de tercera generación como el paclitaxel, ha alcanzado respuestas objetivas del 20% al 35%, con una mediana de sobrevida libre de progresión (SLP) de cuatro a cinco meses y una sobrevida global (SG) de ocho a 11 meses.13 Los regímenes actuales de quimioterapia tienen una eficacia limitada, con un modesto beneficio en términos de sobrevida y conllevan una toxicidad significativa, que da lugar a que muchos pacientes no puedan recibir este tratamiento, incluso en el marco de terapia de primera línea. De acuerdo a esto, en la actualidad existe la necesidad de proporcionar a los pacientes agentes menos tóxicos, como las novedosas terapias dirigidas, con el potencial de mejorar la eficacia y mantener una buena calidad de vida con una baja toxicidad. La inhibición de receptores con actividad de TK mediante la administración de anticuerpos monoclonales, RNAs de interferencia y/o EGFR-TKIs, impiden la proliferación y la supervivencia de células neoplásicas, induciendo el arresto celular y apoptosis. Los tratamientos moleculares contra la vía del EGFR, son una de las estrategias terapéuticas en el CPCNP. Los tratamientos con agentes biológicos tales como los anticuerpos monoclonales y los EGFR-TKIs, son la opción para ser usados como tratamientos de primera y segunda línea, en pacientes con CPCNP avanzado. Puesto que presentan una toxicidad aceptable y han mostrado resultados sorprendes, en un grupo particular de pacientes. Los anticuerpos como el cetuximab,14 se unen competitivamente al dominio extracelular del EGFR inhibiendo la asociación de su ligando e impidiendo su dimerización, fosforilación y activación. Además, la inhibición del receptor induce su baja regulación y eventualmente su internalización y degradación, proceso que explica la actividad antitumoral de estos anticuerpos.15
¿ INHIBIDORES DE CINASAS DE TIROSINAS (TKIs)
Los TKIs son moléculas pequeñas que inhiben la actividad enzimática de los receptores de factores de crecimiento. Muchas de estas moléculas, pueden categorizarse en los siguientes grupos: 1) Inhibidores competitivos del ATP, los cuales se unen predominantemente al sitio de unión del ATP de la cinasa, cuando este sitio está en la conformación activa. 2) Inhibidores que reconocen y se unen al sitio de unión del ATP en su forma inactiva y de esta manera haciendo energéticamente desfavorable la activación. 3) Inhibidores alostéricos los cuales, se unen fuera del sitio de unión del ATP pero modifican la estructura tridimensional del receptor evitando la interacción entre el ATP y el dominio de TK. 4) Inhibidores covalentes, que se unen irreversiblemente al sitio de unión del ATP de la cinasa. Los TKIs son efectivos contra blancos moleculares membranales o intracelulares y han sido los métodos de inhibición del EGFR, más exitosos.16
El marcador más usado y confiable para la selección de pacientes candidatos a ser tratados con EGFR-TKIs, es la detección de mutaciones en los exones 19 al 21 del EGFR. Desafortunadamente, se ha observado que el 50% de los pacientes tratados con EGFR-TKIs desarrollan resistencia adquirida después de seis a 12 meses de tratamiento con dichos agentes, debido a que se genera la mutación T790M en el exón 20 del EGFR. Esta mutación reduce hasta cien veces la capacidad inhibitoria de los EGFR-TKIs. Los estudios de modelado estructural han postulado que la mutación T790M, genera un incremento de unión del ATP al EGFR por más de un orden de magnitud, lo que facilita la fosforilación del EGFR, especialmente cuando se encuentra en conjunto con la mutación L858R, lo que origina la resistencia al gefitinib o erlotinib.5
¿ ERLOTINIB Y GEFITINIB, INHIBIDORES DE LA ACTIVACIÓN DEL EGFR
El erlotinib y el gefitinib son moléculas pequeñas que funcionan como inhibidores del dominio intracelular con actividad de TK del EGFR, se unen de manera reversible a este dominio compitiendo con el ATP e inhibiendo la transfosforilación de los residuos de tirosinas del EGFR y bloquean las señales de transducción.17
Actualmente, tanto el erlotinib como el gefotinib son utilizados como terapia molecular contra el cáncer, porque reduce la autofosforilación del EGFR en células tumorales, bloquea la progresión de la fase G1 del ciclo celular e induce apoptosis.18 Los efectos adversos más comunes asociados al tratamiento, son diarrea y rash. Sin embargo, otras toxicidades frecuentes son: dolor de cabeza, naúsea, fatiga, incremento transitorio de bilirrubina sérica y transaminasa.17
La FDA (Food and Drug Administration) aprobó el uso de erlotinib, en combinación con gemcitabina como primera línea de tratamiento de pacientes con cáncer de páncreas localmente avanzado, irresecable o metastásico.
Lo anterior basado en un estudio clínico fase III, que mejoró la supervivencia libre de progresión (SLP) y supervivencia global (SG) de los pacientes con este tipo de tumor.19 El erlotinib también está en investigación para su uso en el tratamiento de otro tipo de tumores, incluyendo el cáncer de mama, cabeza y cuello, colon y ovario en combinación con quimioterapia y otras terapias moleculares dirigidas.16
La eficacia y la seguridad del erlotinib se evaluaron recientemente, en pacientes con glioblastoma que tuvieron una primera recaída.20 Los resultados de este trabajo mostraron medianas de respuestas de siete meses, 20% de SLP a seis meses y una mediana de SG, de 9.7 meses. El 23% de los pacientes mostraron efectos adversos grado tres a cuatro relacionados al tratamiento. La diarrea y rash fueron los efectos más comunes. De manera interesante las respuestas no estuvieron asociadas a la amplificación del gen EGFR.20
¿ ERLOTINIB Y GEFITINIB COMO TRATAMIENTO DEL CPCNP
El erlotinib ha mostrado considerables índices de respuesta, en estudios clínicos fase II en pacientes con CPCNP avanzado, que recibieron tratamiento previo. Basado en un estudio clínico randomizado fase III,21 el erlotinib fue aprobado por la FDA para el tratamiento de CPCNP localmente avanzado o metastásico, que haya fallado al menos a una primera línea de quimioterapia. El estudio internacional BR.2121 aleatorizó 731 pacientes con CPCNP avanzado, que fallaron previamente a uno o dos regímenes de quimioterapia a recibir erlotinib o placebo (estudio NCIC-CTG BR.21). Tanto la respuesta al tratamiento (8.9% vs <1%, p<0.001), la SG (6.7 vs 4.7 meses, p<0.01) y la calidad de vida de los pacientes, mejoró significativamente en el grupo que recibió erlotinib.21 El género femenino, el origen asiático sin historia de tabaquismo y el adenocarcinoma se asociaron con la respuesta, pero la única característica clínica que se asoció con un beneficio en la supervivencia fue la ausencia de historia de tabaquismo. Las tasas de respuesta y tolerancia a erlotinib fueron confirmadas en 6 708 pacientes, dentro del estudio TRUST.22 Los análisis multivariados que evalúan características clínico patológicas sugieren, que los pacientes con adenocarcinomas, sin antecedentes de tabaquismo y con expresión de EGFR correlacionan con las respuestas al erlotinib.23 Sin embargo, en el estudio BR.21, la expresión, el número de copias y el análisis de mutaciones del EGFR no muestran una asociación significativa con la supervivencia de los pacientes, cuando se hacen análisis multivariados pero sugieren estudios prospectivos.24
Como tratamiento de primera línea para CPCNP avanzado, la adición de erlotinib a la quimioterapia estándar basada en platinos no tiene beneficio en términos de sobrevida.25,26 No obstante, estos trabajos sugieren que la supervivencia puede aumentar, cuando se administra erlotinib a los pacientes que tuvieron una enfermedad estable o respondieron a la quimioterapia (Tabla 1). Un estudio fase II en pacientes con carcinoma bronquioalveolar que recibieron erlotinib, mostraron una respuesta de 25% que correlacionó con tabaquismo negativo y con mutaciones en EGFR y no hubo respuesta en pacientes con mutaciones en KRAS.27 En otro estudio fase II, en el cual se usó erlotinib en 150 pacientes con CPCNP, reportó una respuesta que correlacionó con las características clínicas asociadas que fueron: adenocarcinoma y la exposición al humo de leña. Lo que les permitió concluir que la exposición al humo de leña está asociada con una mejor respuesta al erlonitib, mejorando la progresión libre de enfermedad en pacientes con CPCNP.28
El gefitinib ha mostrado ser efectivo como primera línea de tratamiento en pacientes asiáticos, con CPCNP avanzado y con características clínicas favorables como el género femenino, sin antecedentes de tabaquismo y con adenocarcinoma.29,30 Un estudio aleatorizado, doble ciego, controlado con placebo, en grupos paralelos, multicéntrico, fase III, Evaluación de Sobrevida en Cáncer Pulmonar con GEFITINIB (ISEL, por sus siglas en inglés), se diseñó para investigar el efecto de sobrevida de gefitinib 250 mg/día más el tratamiento paliativo, en pacientes con CPCNP que eran refractarios o intolerantes a su esquema de quimioterapia más reciente. El punto final primario de ISEL, fue la supervivencia en las poblaciones de pacientes en general y con adenocarcinoma. Los puntos finales secundarios incluyeron tiempo hasta el fracaso del tratamiento (ThFT), tasa de respuesta objetiva (TRO), CdV y mejoría de los síntomas y seguridad.31
Los pacientes elegibles tenían >18 años de edad, habían recibido uno o dos esquemas previos de tratamiento para CPCNP y eran refractarios o intolerantes al esquema más reciente de quimioterapia. Para pacientes de <70 años de edad (pero no pacientes >70 años de edad), esto debió contener por lo menos un esquema con platino. Los pacientes fueron clasificados en un principio de acuerdo con la histología del tumor, sexo, antecedentes de tabaquismo, número de esquemas previos de quimioterapia, razón para el fracaso de la quimioterapia previa y ED. Se agregaron los siguientes subgrupos adicionales de pacientes, al análisis de subgrupo estadísticamente riguroso: tratamiento previo con docetaxel, edad al momento de la aleatorización, tiempo desde el diagnóstico al tratamiento, origen racial y mejor respuesta a la quimioterapia previa. Los pacientes fueron aleatorizados en una proporción 2:1 para recibir gefitinib o placebo, además de tratamiento paliativo, de acuerdo con las prácticas locales. En una mediana de seguimiento de 7.2 meses (rango tres a 15 meses), ocurrieron 976 muertes en el estudio ISEL y la tasa total de mortalidad fue de 58%. En la población general, la mejoría en la sobrevida con gefitinib no alcanzó significancia estadística en comparación con placebo (Razón de riesgos de rangos logarítmicos [RR] 0.89; IC del 95% 0.77, 1.02; p=0.087). No obstante, un análisis de riesgos proporcionales de Cox sugirió significancia estadística a favor de gefinitib (RR 0.86; 95% IC del 0.76, 0.99; p=0.030). La mediana de la sobrevida en la población general fue de 5.6 meses para gefitinib, en comparación con 5.1 meses para placebo. Las tasas estimadas de sobrevida a un año para gefitinib y placebo fueron de 27% y 21%, respectivamente en la población en general.31
El estudio TRUST sugiere que la tasa de respuesta y la seguridad en pacientes con CPCNP no seleccionados, son similares tanto en primera, segunda y tercera línea de quimioterapia.32 Hasta la fecha, no se han realizado estudios comparativos fase III entre pemetrexed, docetaxel vs erlotinib, pero analizando las evidencias actuales, los expertos indirectamente infieren que para tratamientos de segunda línea el erlotinib es tan efectivo como el docetaxel en términos de sobrevida. A pesar de la carencia de estudios comparativos, el pemetrexed podría ser al menos tan efectivo como el docetaxel y el erlotinib tan efectivo como ambas drogas.
En tratamientos de primera línea, la toxicidad de la quimoterapia es considerada superior a la del erlotinib y en tratamientos de tercera línea, el erlotinib es el tratamiento más efectivo. Sin embargo, para los tratamientos de segunda línea una selección cuidadosa de los pacientes es requerida. Considerando la eficacia, si se concluye que estas drogas son similares, la decisión para administrarse podría tomarse de acuerdo a aspectos individuales para cada caso. Por ejemplo: en un fumador sin comorbilidades, probablemente la quimioterapia es preferible. En contraste, en una mujer con adenocarcinoma, sin historia de fumadora el tratamiento con erlotinib es la opción más factible puesto que el erlotinib parece beneficiar a estos subgrupos.33
Las evidencias muestran que las opciones para tratamientos de segunda línea, parecen ser comparables en lo que concierne a respuesta. Para seleccionar la terapia más conveniente, los expertos evalúan las condiciones para cada caso junto con el paciente, quien está bien informado de la toxicidad de cada droga. Los datos parecen sugerir que el perfil de seguridad del erlotinib, es más favorable que el de drogas de uso general de segunda línea. En estudios internacionales aplicados en otros países, hacen comparaciones acerca de la rentabilidad del uso del erlotinib, con las otras dos opciones para el tratamiento de segunda línea, los cuales muestran ventajas más altas para el uso del erlotinib. En América Latina, se hicieron dos estudios donde analizaron la rentabilidad y consideraron no sólo la medicación y costo del servicio médico sino también, el costo de las complicaciones que pueden presentarse en cada administración, generalmente neutropenia por quimioterapia y diarrea o rash por erlotinib. De acuerdo, a la investigación realizada con docetaxel y pemetrexed que consideró todos los costos hasta medicación adicional y principalmente costos de hospitalización, los costos de docetaxel fueron mucho más altos a comparación de los de pemetrexed.33 No hay conocimiento de análisis de rentabilidad del erlotinib, realizados por instituciones locales. Sin embargo, una empresa consultora independiente realizó un análisis local centrado en mujeres con adenocarcinoma y comparó la rentabilidad del erlotinib con la del docetaxel y demostró una ventaja de rentabilidad para el erlotinib.32 Por lo tanto, el erlotinib es una opción efectiva para ser usado como tratamiento de segunda línea.
¿ PERSONALIZACIÓN DE TRATAMIENTO
Los datos recientes acerca de la selección molecular de los pacientes con mutaciones en el EGFR que favorecen la respuesta a erlotinib, han mejorado el conocimiento acerca del uso adecuado de estos agentes. Un estudio prospectivo de la determinación de mutaciones en el EGFR, fue llevado a cabo por el grupo de cáncer de pulmón en España.34 Dos mil ciento cinco pacientes con CPCNP avanzado fueron evaluados, de estos 350 fueron positivos a mutaciones en el EGFR (16.6%), la mutación más frecuente fue L858R en el exón 19 (62.2% vs 37.8%). Las mutaciones en EGFR fueron más comunes entre mujeres (69.7%), no fumadores (66.6%) y con adenocarcinomas (80.9%). Doscientos diecisiete pacientes con mutaciones en EGFR fueron tratados con erlotinib, los cuales tuvieron una SLP y SG de 14 y 27 meses, respectivamente. En el análisis multivariado se encontró una asociación entre una SLP pobre, género masculino (HR 2.94, 95% CI 1.72-5.03, p<0.001) y la presencia de la mutación L858R (HR 1.92, 95% CI 1.19-3.10; p=0.02).34
Aunque el tabaquismo es considerado el principal factor de riesgo para desarrollar CPCNP, otros factores de riesgo como la exposición al humo de leña tienen gran importancia en el desarrollo de esta neoplasia. El humo de leña es un carcinógeno humano, pero se desconoce su mecanismo de acción en la carcinogénesis pulmonar. El cáncer pulmonar asociado a tabaquismo con respecto al asociado al humo de leña, presenta comportamientos clínicos distintos con características moleculares y biológicas únicas y específicas. Esto sugiere que los tumores pulmonares asociados a la exposición al humo de leña, podrían tener un patrón de expresión genética distinto al de los tumores asociados a tabaquismo. Nuestro grupo de investigación, encontró mayores tasas de respuesta al tratamiento con erlotinib, comparado con lo reportado a nivel internacional (34% vs 9%), en pacientes con CPCNP avanzado refractario a quimioterapia,33 lo que sugiere una mayor frecuencia de mutaciones de EGFR en nuestra población.
¿ GENOTIPIFICACIÓN DE PACIENTES CON CPCNP EN EL INCAN
Nuestro grupo de trabajo ha determinado el estatus de mutaciones de los genes EGFR y KRAS, de 381 pacientes con CPCNP que acuden al INCan, con la finalidad de proveer el tratamiento adecuado al paciente oncológico y mejorar su calidad de vida. A dichos pacientes, se les tomó la biopsia por tru-cut y posteriormente se aisló el DNA, para finalmente detectar mutaciones en los genes mencionados previamente, mediante PCR en tiempo real. Los resultados fueron los siguientes: de 381 pacientes evaluados, 191 (31.2%) presentaron mutaciones en el EGFR de los cuales, 11 (9.2%) mostraron mutaciones en el exón 18, 76 (63.9%) presentaron deleciones en el exón 19, 31 (26%) la mutación L858R en el exón 21,10 (8.4%) mostraron la mutación S768I en el exón 20, 11 (9.2%) albergaron mutaciones complejas, mientras que únicamente ocho (2.1%), pacientes presentaron la mutación T790M. Todas las mutaciones mencionadas anteriormente, presentaron una asociación independiente con la histología de adenocarcinoma, la edad avanzada y ausencia de historia de tabaquismo.
La presencia o ausencia de mutaciones en el gen KRAS, se evaluó en solo 202 pacientes, de los cuales 34 (16.8%) contuvieron la mutación. Aunque la mayoría de los informes indican que las mutaciones en EGFR y KRAS se excluyen entre sí, nuestros resultados muestran que mutaciones del gen KRAS pueden coexistir con mutaciones en el EGFR, similar a un reporte anterior.35 Nuestros resultados sugieren una mayor frecuencia de mutaciones del gen KRAS en fumadores que en no fumadores, como ha sido mostrado en reportes anteriores.36 Pacientes con mutaciones en EGFR y ausencia de mutaciones en KRAS, recibieron tratamiento a base de terapias biológicas con EGFR-TKIs en primera, tercera y cuarta línea de tratamiento. Los resultados clínicos fueron: respuesta completa 7,1%, respuesta parcial el 55,4% (tasa de respuesta global del 62,5%) y enfermedad estable en el 37,5%. SLP y SG fueron de 15,1 (IC 95%: 12.4-17.9) y de 16,4 meses (12.4-20.6), respectivamente. No se encontraron diferencias en la SG entre los pacientes portadores de deleciones en el exón 19 (16,5 meses, 10.4-22.7) o L858R (16 meses, 11.1-20.9, p=0,612), resultados similares fueron reportados en pacientes tratados con gefitinib como terapia de primera línea en pacientes con mutaciones en EGFR.37 Es importante destacar, que únicamente ocho (2.1%) pacientes presentaron la mutación T790M, esta mutación se genera posterior al tratamiento con EGFR-TKIs y esta reportada como una mutación de resistencia adquirida a estos inhibidores.5 No obstante, existe ya en el mercado un nuevo EGFR-TKI denominado afatinib, el cual es un medicamento que tiene la capacidad de unirse y bloquear al EGFR a pesar de la presencia de la mutación de resistencia T790M.38 Este nuevo EGFR-TKI, está siendo investigado en el programa de LUX-Lung, que evalúa a el afatinib como tratamiento de primera línea en pacientes con mutaciones que activan el EGFR (LUX-Lung 2, 3 y 6) y como tratamiento de segunda o tercera línea en pacientes que han adquirido resistencia al gefitinib y / o erlotinib (LUX-Lung 1, 4 y 5). LUX-Lung 1 y 2 han demostrado, dentro de sus grupos respectivos, un aumento significativo en la tasa de control de la enfermedad de 58% y 86%, respectivamente, y una prolongación significativa de supervivencia libre de progresión. Además de fase III de ensayos clínicos, actualmente en curso para evaluar afatinib en combinación con paclitaxel (LUX-Lung 5), y en comparación con cisplatino/ pemetrexed (LUX-Lung 3) o cisplatino / gemcitabina (LUX-Lung 6).
Respecto a esto, queremos recalcar la importancia de la genotipificación de los pacientes con CPCNP, la cual tiene un impacto importante para determinar el tratamiento adecuado, con la finalidad de mejorar la calidad de vida del paciente oncológico.
¿ MANTENIMIENTO CON ERLOTINIB
La terapia de mantenimiento en pacientes con CPCNP después de cuatro a seis ciclos de quimioterapia, puede retrasar la progresión de la enfermedad y prolongar la supervivencia. Cappuzzo y colaboradores llevaron a cabo un estudio fase fase III, que usó erlotinib como terapia de mantenimiento en pacientes con CPCNP irresecable y con enfermedad no progresiva, después de la primera línea de tratamiento con quimioterapia a base de platino. Ochocientos ochenta y nueve pacientes que no progresaron a la enfermedad, fueron randomizados a recibir erlotinib 150 mg/día o placebo hasta la progresión o toxicidad inaceptable. Los pacientes fueron estratificados por estatus de EGFR, estadio, ECOG, régimen de quimioterapia, tabaquismo y región. Después de una mediana de seguimiento de 11.4 meses del grupo de erlotinib contra 11.5 del grupo placebo, se evaluó la SLP. En el grupo de erlotinib fue de 12.3 semanas contra 11.1 semanas en el grupo placebo (HR 0.71 95% CI 0.62-0.82; p<0.001). La SLP fue más alta en pacientes con sobreexpresión de EGFR y tratados con erlotinib (12.3 semanas), comparado contra los que sobreexpresan EGFR y recibieron placebo (11.1 semanas) (HR 0.69, 0.58-0.82; p<0.001). Los eventos adversos grado tres, más comunes fueron rash y diarrea. Serios eventos adversos se reportaron en 47 pacientes (11%) del grupo de erlotinib y en 34 (8%) del grupo placebo. El evento adverso más común fue neumonía (siete casos en el grupo de erlotinib 2% y cuatro < 1%, en el grupo placebo).39 Este estudio sugiere que la terapia de mantenimiento con erlotinib es bien tolerada en pacientes con CPCNP avanzado y prolonga significativamente la SLP. Además, puede ser utilizada en pacientes que no progresaron después de cuatro ciclos de quimioterapia.39
¿ FUSIÓN DEL ONCOGÉN EML4-ALK
En el linfoma anaplásico de células grandes, el gen de la cinasa del linfoma anaplásico (ALK) se une con el gen de nucleofosmina (MNP), generando una proteína de fusión NPM-ALK, la cual juega un papel esencial en la linfomagenésis. Aunque la unión de ALK se ha relacionado con neoplasias hematológicas, en el 2007 se describió una nueva fusión asociada a NSCLC que involucra a ALK, con el gen de la proteína 4 asociada a microtúbulos equinodermos (EML4). La fusión resultante genera una inversión pequeña dentro del cromosoma 2p; la inv. (2) (p21; p23), la cual conduce a la expresión de una tirosina-cinasa quimérica, que induce crecimiento constitutivo de las células, por lo tanto, la fusión EML4-ALK posee actividad oncogénica tanto in vitro como in vivo. Diversos estudios de pacientes, han reportado una frecuencia de expresión entre un 3% y 13% de la fusión EML4-ALK. Además las características clínicas asociadas a esta mutación, son muy similares a los pacientes que presentan mutaciones en el receptor del EGFR, estos pacientes parecen ser más jóvenes que el paciente promedio de cáncer de pulmón (edad media 52 años), no fumadores y con predominancia histológica de adenocarcinomas (principalmente en adenocarcinoma con células en anillo de sello). Cabe mencionar, que los pacientes con EML4-ALK no presentan mutaciones de EGFR y del gen KRAS. Por otra parte, dependiendo del punto de ruptura del gen EML4 existen variantes o isoformas de la fusión, las cuales se han confirmado por diferentes grupos de investigaciones (Figura 3).
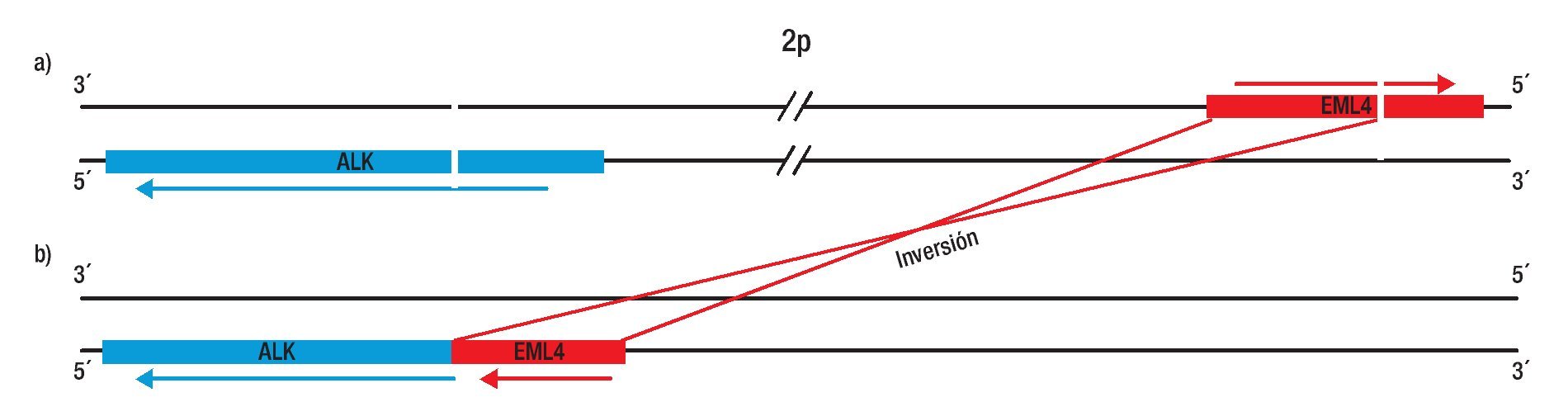
Figura 3. Representación esquemática del mecanismo de fusión de ALK con EML4: Inv. (2) (p21p23). Gen de EML4 y de ALK a), Inversión de EML4 y ALK generando la fusión EML4-ALK b).
En México, poco se conoce sobre la frecuencia de esta nueva alteración citogenética en pacientes con cáncer de pulmón. El impacto de la predicción sobre la frecuencia del gen de expresión de EML4-ALK, en diferentes poblaciones étnicas es motivo de estudio. Las técnicas de RT-PCR o FISH son métodos sensibles y fiables para la detección de tumores con células EML4-ALK positivos. Es un criterio de diagnóstico la determinación de EML4-ALK, para tomar decisiones respecto al tratamiento quimio-terapeútico con inhibidores específicos. El inhibidor de la molécula de ALK (PF-02341066-Crizotinib), ha mostrado tener buenos resultados en pacientes con cáncer de pulmón positivo para la translocación ALK, los cuales conducen a la muerte celular vía apoptosis in vitro y a la reducción del tumor in vivo. Los pacientes tratados con crizotinib presentan respuestas alrededor de 57% y control de la enfermedad a seis meses del 72%. Sin embargo, al igual que mutaciones de EGFR en T790R, confiere resistencia a ITK, se han descubierto adquiridas mutaciones dentro del dominio cinasa del gen EML4-ALK, que confiere resistencia al crizotinib.40,41
En conclusión, la genotipificación de pacientes con CPNCP es fundamental para el tratamiento, y debe ser considerado un estándar en el manejo de estos pacientes. Los factores predictivos de respuesta a los Inhibidores de TK son las mutaciones de sensibilidad en el EGFR, como la delección del exon 19 y mutaciones puntuales del exón 21. La frecuencia de este tipo de mutaciones varía de acuerdo al origen étnico, en Latinoamérica parece ser intermedio a asiáticos y a caucásicos.42 La falta de respuesta a estos inhibidores está asociada a la traslocación del gen de ALK y mutaciones en KRAS. Otras mutaciones están siendo estudiadas como la BRAF, la cual que presenta en alrededor de 3% y esta asociada al tabaquismo, inhibidores de BRAF están siendo estudiados actualmente en esta población.43
Correspondencia: Dr. Oscar Arrieta.
Coordinador de la Clínica de Tumores Torácicos, Instituto Nacional de Cancerología de México (INCan). Av. San Fernando No. 22 Col. Sección XVI. C.P. 14080. México, D.F., México.
Teléfono: (52 55) 5628 0400, Ext: 832. Fax: (5255) 551315 1223.
Correo electrónico:ogar@unam.mx