






Es la resistencia que puede generar cualquier organismo patógeno a determinados fármacos o compuestos químicos a los que usualmente son letales para su especie. Es un proceso biológico de carácter genético y heredable entre los miembros de la misma especie. Cuando una población de seres vivos tienen individuos con algún gen que le permite una mayor resistencia a una determinado compuesto tóxico que al resto de la población, ésta resistencia natural puede ejercer una fuerte presión selectiva si se recibe en dosis tales que sea más letal para los que no son resistentes. Los individuos resistentes tendrán más probabilidad de sobrevivir, reproducirse y transmitir el carácter de la resistencia a su descendencia, de modo que el porcentaje de la población resistente a esta dosis particular, se incrementará selectivamente. Las mutaciones sucesivas de los genes responsables de la quimiorresistencia, o la variabilidad genética que permita que solo algunos individuos hereden estos genes que otorgan resistencia al tóxico de forma sinérgica, permitirá la adaptación progresiva de un organismo al tóxico mientras exista esa presión selectiva, lo cual muchas veces logra alcanzar la resistencia total a dosis elevadas del compuesto tóxico.
¿ IMPORTANCIA DE LA QUIMIORRESISTENCIA EN LA CLÍNICA ONCOLÓGICA
La tasa de morbilidad y mortalidad atribuida a tumores malignos por habitante y año son cifras en continuo crecimiento a nivel mundial. La prevención sanitaria contra determinadas sustancias cancerígenas, así como la detección y tratamiento precoz de tumores potencialmente curables han conseguido hasta el momento resultados muy limitados. La falta de respuesta al tratamiento y la recidiva de tumores inicialmente quimiosensibles son responsables de un número importante de muertes en pacientes neoplásicos. Las opciones terapéuticas utilizadas como rescate, tales como la quimioterapia alternante, la intensificación de la dosis o la quimioterapia regional, no han aportado hasta ahora los resultados esperados.1 La necesidad de nuevas alternativas terapéuticas ha estimulado la investigación sobre los mecanismos que provocan la quimiorresistencia en las células tumorales y sobre las sustancias capaces de inhibirla.
¿ RESISTENCIA INTRÍNSECA Y ADQUIRIDA EN EL TRATAMIENTO DEL CÁNCER
La resistencia intrínseca, natural o de novo se ha descrito en tumores que presentan conocida falta de respuesta a la quimioterapia, como ocurre con el hipernefroma, el cáncer de colon, el cáncer de páncreas o el melanoma. La mayor parte de los pacientes con tumores que responden inicialmente a la quimioterapia presentan recaídas en su evolución, insensibles al tratamiento por la adquisición de la llamada resistencia adquirida a múltiples fármacos antineoplásicos (MDR: multidrug resistance).2 La investigación sobre genética bacteriana y sobre los mecanismos de resistencia bacteriana, así como el desarrollo de técnicas con DNA recombinante han desempeñado un papel importante en el conocimiento de los mecanismos de resistencia implicados en las células tumorales. Los fenómenos de mutación y amplificación genética responsables de la resistencia a bacteriófagos en E. coli fueron el punto de partida de la investigación sobre quimiorresistencia en las células tumorales. Goldie y Coldman relacionaron la capacidad de mutación genética propia de estas células con la aparición de resistencia, y definieron los conceptos de resistencia intrínseca y adquirida.3,4 En las primeras experiencias con cultivos de células tumorales, se observó la aparición de quimiorresistencia a medida que se incrementaba la concentración de la sustancia empleada: la actinomicina D. Esta resistencia también afectaba a otro grupo de fármacos como la colquicina, vinblastina o adriamicina, no relacionadas estructural ni funcionalmente con la actinomicina D. En el estudio farmacocinético de estos fármacos se observó un bombeo activo al exterior de la célula cuando se unían a una proteína de membrana plasmática, presente sólo en las células resistentes.1,5,6 Varios trabajos que intentaron aclarar los mecanismos implicados en estos fenómenos de resistencia, obtuvieron resultados similares.7-9 Juliano y Ling demostraron una correlación entre el grado de resistencia a la colchicina en las células tumorales, la presencia de una glucoproteína de 170 kD en la membrana plasmática y la amplificación del gen codificador de dicha proteína, el gen MDR1.5,10-12 La gran similitud de esta glucoproteína con las proteínas transportadoras bacterianas, sobre todo con la hemolisina B y el sistema malk de E. coli y sistema hisP del Staphilococcus typhimurium, sugirió su mecanismo de acción como transportador de membrana.12,13
¿ LA GLUCOPROTEÍNA P (G-P)
Es una proteína de membrana plasmática de 170 kD, formada por 1200 aminoácidos que se disponen en dos cadenas iguales, cada una con un segmento N-terminal hidrofílico y un segmento C-terminal hidrofóbico. Cada cadena dispone de seis regiones transmembrana a las que se pueden unir sustancias naturales del tipo de las vincas, podofilotoxinas, colchicina, actinomicina D y/o antraciclinas. En su cara intracelular hay una región hidrofílica a la que se liga una molécula de ATP, por lo que se piensa que el mecanismo de acción es dependiente de la energía.14-18
¿ FUNCIONAMIENTO DE LA GLUCOPROTEÍNA P
La estructura de la G-P es característica de un poro de membrana a través de la que los diferentes fármacos son bombeados al espacio extracelular, disminuyendo la concentración en el interior de la célula a niveles que resultan inactivos.12-14,18,19 La unión del fármaco a la G-P es esencial para su inactivación (Figura 1).
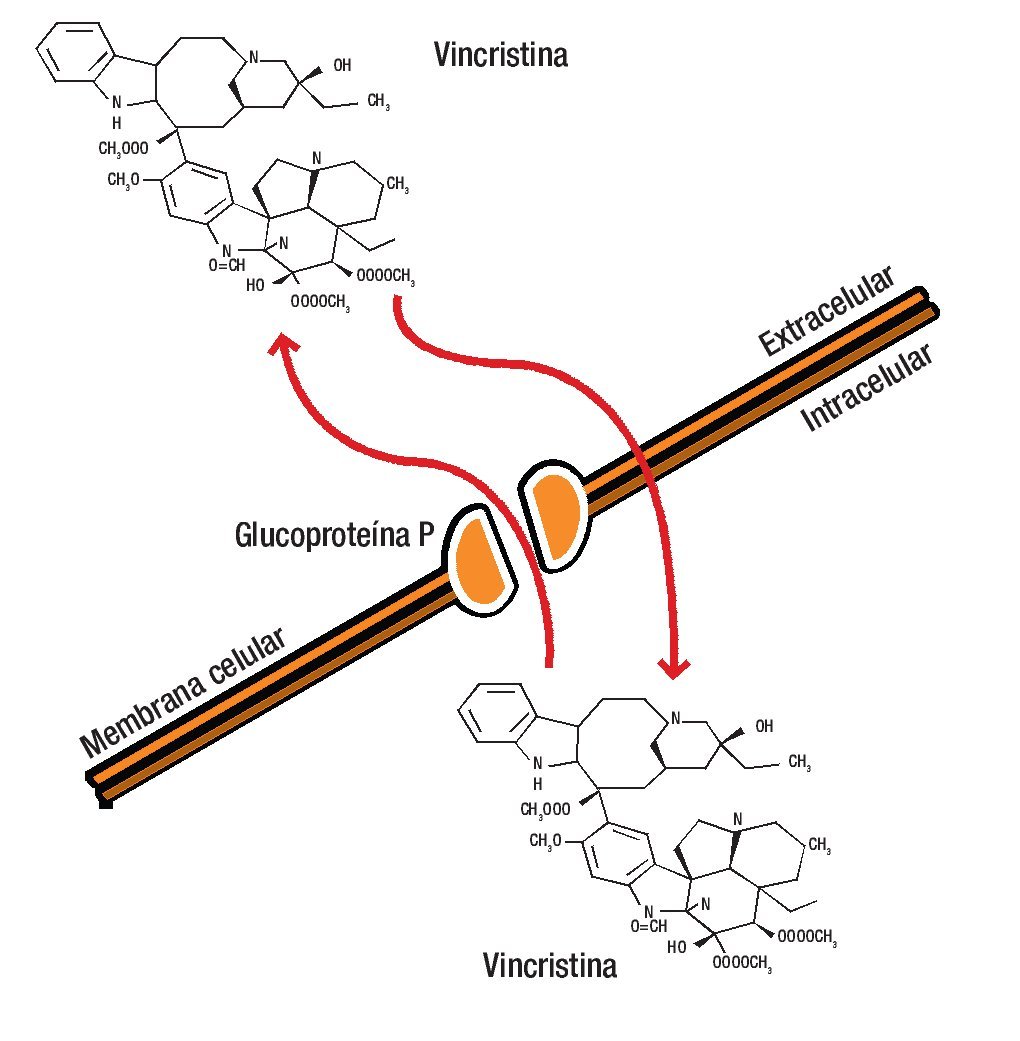
Figura 1. La glucoproteína P es una molécula de la membrana celular que se caracteriza por fromar un poro a través del cual los diferentes fármacos son bombeados al espacio extracelular, disminuyendo así la concentración en el interior de la célula a niveles que resultan inactivos.
La G-P posee varios epítopes o puntos de unión para diferentes sustancias, por lo que puede provocar su resistencia de forma simultánea.1,14 El número de epítopes es limitado y los diferentes fármacos deben competir por su unión con esta proteína. Este hecho ha estimulado la investigación de sustancias que se unan a la G-P con gran afinidad, la bloqueen y de esta forma, mediante un mecanismo de inhibición competitiva, impidan la expulsión de los fármacos al espacio extracelular.20
¿ CARACTERÍSTICAS GENÉTICAS DE LA GLUCOPROTEÍNA P
La G-P está codificada por el gen MDR1. En la especie humana la familia multigénica MDR está constituida por dos genes: MDR1 y MDR2 presentasen el brazo largo (banda q21.1) del cromosoma siete.18,21,22 Ambos genes se transcriben en un RNA de 4,1-4,5 kb, con un alto grado de homología entre ellos. Las proteínas codificadas por ambos genes tienen una orientación similar en las membranas celulares, actuando como transportadoras activas.18 En la actualidad, sólo se ha demostrado la participación del MDR1 y de su producto, la G-P, en los mecanismos de quimiorresistencia.23 Las células con fenotipo MDR presentan invariablemente alteraciones cromosómicas. La amplificación del gen MDR1, demostrada en algunos casos por la presencia de dobles minutos, o la desregulación en la transcripción del MDR1 a RNA-MDR1 son las más frecuentes.7,14 Esta última es la única alteración observada en los tumores humanos quimiorresistentes.15,24 La transferencia del material genético DNA-MDR1 a células sensibles es capaz de inducir en ellas una quimiorresistencia, como se ha observado por técnicas de hibridación en virus.19 El aislamiento de vector retrovírico portador de DNA-MDR1 ha permitido la transferencia de quimiorresistencia a cultivos celulares. Este hecho abre un interesante campo de investigación terapéutica con la posibilidad de proteger contra la aplasia medular posquimioterapia, mediante la transferencia de un vector retrovírico DNA-MDR1 a las células madre de la medula ósea.25,26 La expresión del gen MDR1 en tumores intrínsecamente resistentes, así como su aparición en tumores que adquieren esta resistencia, sugieren su participación en los mecanismos de quimiorresistencia.26
¿ PAPEL FISIOLÓGICO DE LA GLUCOPROTEÍNA P
La G-P es tejido-específica y se detecta, casi de forma exclusiva, en los epitelios de órganos maduros con capacidad excretora-secretora, endotelios y tejido trofoblástico. La corteza adrenal, el túbulo renal proximal y el epitelio colónico expresan G-P de forma difusa y con mayor intensidad que en el resto de tejidos, lo que facilita la eliminación de metabolitos y de diferentes sustancias a la bilis, orina o luz intestinal, y confirma el papel detoxificante y protector celular de la G-P.6,21,27 La detección de G-P en el endotelio capilar del testículo y del sistema nervioso central sugiere su participación en la creación de santuarios para enfermedades como las leucemias, al impedir que los fármacos alcancen concentraciones activas en estas localizaciones.28
¿ LA EXPRESIÓN DE LA GLUCOPROTEÍNA P EN LOS TUMORES HUMANOS
Bell y colaboradores (1985), observaron una correlación entre la presencia de G-P en células de carcinoma ovárico y su grado de quimiorresistencia. Estos resultados, que fueron obtenidos por técnicas de inmunoblot, han sido confirmados en los últimos años mediante RNA slot blot, RNA hibridación in situ, inmunohistoquimia, y PCR.29,30 El análisis del RNA-MDR1 en más de 400 tumores diferentes ha confirmado la alta expresión del gen MDR1 en los que son intrínsecamente quimiorresistentes, como el hipernefroma, el cáncer adrenal, el cáncer de colon o el hepatocarcinoma.30-32 La variabilidad en el patrón de distribución, así como en la intensidad de expresión de la G-P en los diferentes tumores ha sido demostrada por técnicas de inmunohistoquimia, utilizando anticuerpos monoclonales dirigidos a diferentes epítopes de la G-P (C219, MRKl6, JSB1, entre otras). De la tejidos que no expresan G-P en condiciones normales, lo que apoya la teoría de una resistencia adquirida.6,18,33-38 Uno de los puntos que más interés ha despertado en el conocimiento de la G-P es el papel que desempeña como factor pronóstico y como predictor de respuesta tumoral a la quimioterapia. En los recientes años se han publicado varios trabajos en los que se demuestran una correlación entre la presencia de G-P en las células tumorales con una mayor agresividad del tumor, es decir, con mayor invasividad vascular, mayor afectación ganglionar y diseminación a distancia, así como un intervalo libre de enfermedad más corto y una menor respuesta tumoral al tratamiento.14,26,34-42 La ausencia de G-P en las células tumorales no es predictiva de la eficacia terapéutica ya que otros mecanismos de resistencia pueden estar presentes. Han sido implicados varios sistemas enzimáticos en la falta de respuesta a determinados fármacos, principalmente el sistema de la glutatión-S-transferasa en el citoplasma y el de la topoisomerasa endonucleasa en el núcleo.43,44
¿ AGENTES MODIFICADORES DE LA QUIMIORRESISTENCIA
En un intento de mejorar los resultados terapéuticos se han realizado un importante número de trabajos con el objetivo de bloquear los mecanismos que provocan quimiorresistencia. Gottesman consiguió inhibir la resistencia a diferentes fármacos como la adriamicina, la vinblastina, el etopósido o la actinomicina D mediante el empleo de una sustancia moduladora, el verapamilo.45 El verapamilo tiene una gran afinidad por la G-P, compite con las sustancias activas por los diferentes epítopes, los bloquea y de esta forma inhibe su bombeo al espacio extracelular, es decir, revierte la quimiorresistencia.12 Su mecanismo de acción no está bien definido; se cree que es independiente del calcio y que actúa tanto en la membrana plasmática como en el núcleo, modulando el daño celular provocado por los fármacos, independientemente de su concentración intracelular. En algunos tumores como el carcinoma microcítico de pulmón se ha descrito una acción citotóxica directa. Pese a que el mecanismo de acción no está bien establecido, se han desarrollado un gran número de experiencias con el verapamilo, con la finalidad de mejorar la eficacia del tratamiento antineoplásico. Dalton y colaboradores,37 combinaron el verapamilo con la quimioterapia en un grupo de mielomas y LNH resistentes al tratamiento. La concentración intracelular de los antineoplásicos aumentó en 40%. En otro estudio se obtuvo 72% de respuestas (28% RC) al combinar verapamilo a la quimioterapia (CHOP) en 18 LNH refractarios inicialmente al tratamiento con CHO.14 En estudios in vitro se ha observado una relación dosis-respuesta para el verapamilo, con máximo efecto a concentraciones que provocan una cardiotoxicidad importante, con hipotensión bloqueo cardíaco, que requiere monitorización en UCI.46,47 La presencia de G-P en epitelios normales con capacidad secretora-excretora (renal, colon, hepático, sistema nervioso central) es responsable del aumento de la toxicidad posquimioterapia en estos tejidos cuando se emplea el verapamilo.48 Actualmente está en estudio el papel modulador de diferentes sustancias potencialmente activas y menos tóxicas.
¿ MÚLTIPLE RESISTENCIA A DROGAS (MDR) NO GLUCOPROTEÍNA P
El fenotipo celular MDR se atribuye a una alteración genética causante del incremento de determinadas proteínas, que por su función, inhiben la acción de determinados fármacos. Las proteínas implicadas más conocidas son la G-P en la membrana plasmática, la glutamil-S-transferasa en el citoplasma y la topoisomerasa en el núcleo celular.1 Las alteraciones cualitativas o cuantitativas de enzimas como la topoisomerasa-II se asocian a quimiorresistencia. Esta enzima interviene en la replicación, recombinación y transcripción del DNA celular. La adriamicina, la daunorrubicina, ellipticina, VP16, AMSAM, actinomicina, mitoxantrona entre otras, actúan sobre esta enzima formando un complejo DNA-topoisomerasa II-fármaco altamente estable que bloquea la replicación y la transcripción del DNA y, por lo tanto, provoca la muerte celular. La ausencia de esta enzima en las células tumorales, o la pérdida de la afinidad por los diferentes antineoplásicos provocan quimiorresistencia.21,24 La topoisomerasa-lI es ciclo-específica y presenta las concentraciones más altas en las células de rápido crecimiento. Por este motivo, las células de crecimiento lento pueden tener resistencia al grupo de quimioterápicos que actúan específicamente sobre esta enzima.49 La actividad en el núcleo de la alquiltransferasa, endonucleasa, glucosilasa o de la poli-ADP-ribosa-polimerasa, enzimas reparadoras del DNA, se ha correlacionado con la resistencia a antineoplásicos como las nitrosureas. En el citoplasma existen varios sistemas enzimático detoxificantes celulares, como la glutamil-S-transferasa (GST), superóxido-dismutasa y catalasa que están involucrados en la resistencia a múltiples fármacos.24 La GST promueve el secuestro de la sustancia activa en el interior de endosomas citoplasmáticos protegiendo a la célula de su efecto oxidativo. Las concentraciones de GST son más altas en tejido tumoral que en tejido sano, lo que favorece la resistencia de las células tumorales al tratamiento. El incremento del glutatión intracelular por la acción de la gamma-glutamil-sintetasa y la glutatión-sintetasa, así como el aumento de la actividad óxido-reductora intracelular por la glutatión-peroxidasa y la glutatión-reductasa, se correlaciona con la falta de respuesta a fármacos como el melfalán, el cisplatino o la adriamicina, entre otros. En la actualidad, se investiga la modulación de la MDR no GP a través del empleo de sustancias, como la butioninsulfoximina (BSO), inhibidoras de estos sistemas enzimáticos.50,51
¿ MANEJO DE PACIENTES CON CÁNCER DE MAMA RESISTENTES A TAXANOS
En la actualidad se ha avanzado en el diseño de nuevas drogas y en los esquemas de tratamiento (combinación de drogas, dosis, intervalos de administración, etcétera). Lamentablemente la quimioterapia en muchos casos fracasa porque el tumor presenta mecanismos de resistencia a las drogas administradas (resistencia innata), o los desarrolla una vez iniciado el tratamiento (resistencia adquirida). Los mecanismos de resistencia pueden ser extra o intracelulares; dentro de los mecanismos extracelulares podemos mencionar la irrigación tumoral insuficiente que impide que la droga acceda a la célula tumoral en una concentración adecuada para tener efecto. En cuanto a los mecanismos intracelulares, que son múltiples, los podemos clasificar con fines didácticos en seis grupos. La molécula más estudiada hasta la fecha es la proteína p170 llamada así porque su peso molecular es de 170 kDa (Figura 2).
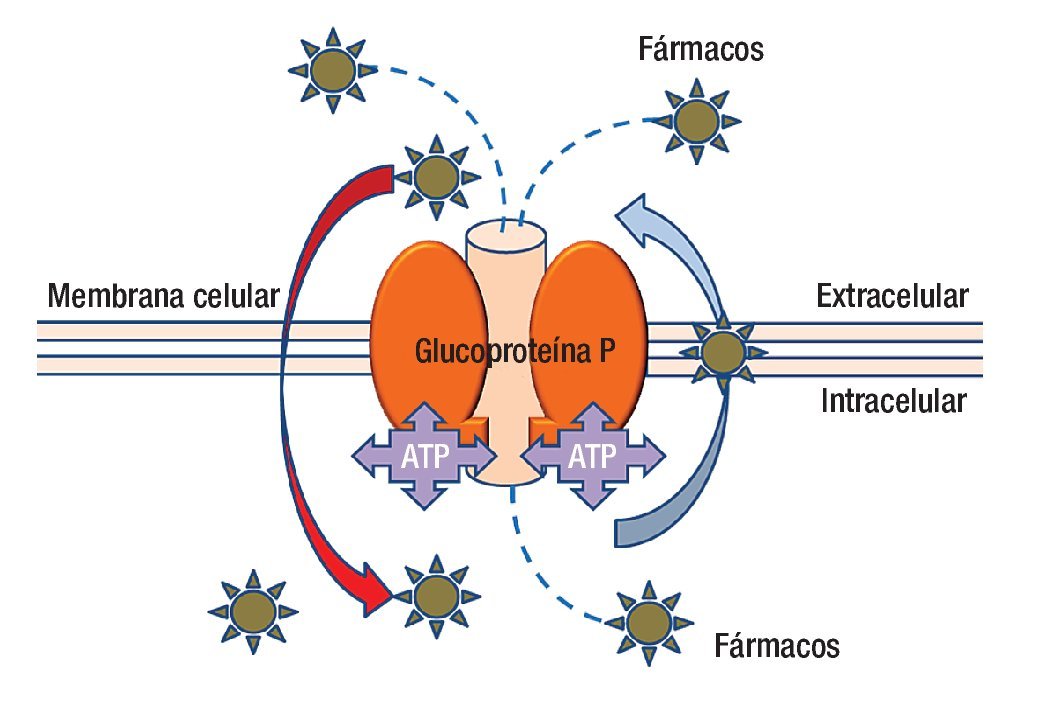
Figura 2. La glucoproteína P es una molécula que anclada a la membrana que además de estar en contacto con ella, tiene presencia dentro y fuera de la célula. La parte central de la molécula es un canal que funciona con energía (ATP) para transportar fármacos del interior de la célula al exterior, incluso puede movilizar fármacos desde el interior de la membrana celular. La polaridad de los agentes farmacológicos es parte de la clave para capturarlos y expulsarlos del interior de la célula.
Esta proteína, que se ubica en la membrana celular, actúa como una bomba que expulsa sustancias nocivas desde el interior de las células hacia el exterior, participando de esta manera en procesos de detoxificación. Forma parte entonces del mecanismo uno (menor acumulación intracelular de las drogas). La p170 se expresa en condiciones normales en la membrana plasmática de las células de numerosos órganos, siendo muy abundante en el riñón, el hígado y el intestino grueso; en cambio su expresión es baja en la glándula mamaria normal. La p170 puede expresarse en altos niveles en tumores de mama y de esta manera conferir resistencia a numerosas drogas antineoplásicas porque es capaz de expulsarlas de las células tumorales impidiendo que actúen y eliminen a dichas células. Hay numerosos trabajos publicados sobre la expresión de la p170 en cáncer de mama y sobre su efecto en la respuesta a la quimioterapia, pero todavía hay controversias con respecto a su utilidad clínica. Otra molécula que puede estar expresada en las células tumorales mamarias y que se ha relacionado con sensibilidad/resistencia a drogas antineoplásicas es la proteína c-erbB-2, también conocida como HER-2/ neu. Esta proteína no se encuentra en glándula mamaria normal y cuando aparece en un cáncer de mama se relaciona con un comportamiento tumoral más agresivo que se reflejará en mayor probabilidad de aparición de metástasis y en una sobrevida menor de la mujer. Esta proteína se localiza en la membrana celular y forma parte de una familia de receptores de factores de crecimiento cuyo miembro más conocido es el Receptor del Factor de Crecimiento Epidérmico (EGFR: Epidermal Growth Factor Receptor). Por su ubicación en la membrana celular y por expresarse exclusivamente en células tumorales es un excelente blanco de nuevas terapias, aunque todavía no se conoce exactamente cuál es su mecanismo de acción. Actualmente, uno de los más novedosos tratamientos en cáncer de mama es la administración por vía sistémica de un anticuerpo monoclonal contra la proteína c-erbB-2: trastuzumab (Herceptin®). Este tratamiento sólo se puede proporcionar a mujeres, cuyo tumor sobre-expresa la proteína c-erbB-2.
¿ AGENTES ESTABILIZADORES DE MICROTÚBULOS
Los microtúbulos son estructuras tubulares de 25 nm de diámetro exterior y unos 12 nm de diámetro interior, con longitudes que varían entre unos pocos nanómetros a micrómetros, que se originan en los centros organizadores de microtúbulos y que se extienden a lo largo de todo el citoplasma. Se hallan en las células eucariotas y están formadas por la polimerización de un dímero de dos proteínas globulares, la alfa y la beta tubulina. Los microtúbulos son heteropolímeros de a y b-tubulina, los cuales forman dímeros, que son su unidad estructural. Son de tipo vegetal. Los dímeros polimerizan en protofilamentos, que luego se agregan lateralmente para formar estructuras cilíndricas huecas. Para polimerizar se requiere la presencia de dímeros a una concentración mínima determinada denominada concentración crítica, aunque el proceso se acelera por la adición de núcleos, que son elongados. Una importante característica de los microtúbulos es su polaridad. La tubulina polimeriza por adición de dímeros en uno o ambos extremos del microtúbulo. La adición es por unión cabeza con cola, en la formación de los protofilamentos. Así, se forman filas sesgadas de monómeros de a y b-tubulina en la pared, lo que provoca una polaridad global al microtúbulo. Debido a que todos los protofilamentos de un microtúbulo tienen la misma orientación, un extremo está compuesto por un anillo de a-tubulina (denominado extremo negativo) y, el opuesto, por un anillo de b-tubulina (denominado extremo positivo). Existen una gran cantidad de drogas capaces de unirse a la tubulina, modular su estado de activación y de este modo interferir con la dinámica microtubular a concentraciones intracelulares, mucho más bajas que la de tubulina. De este modo detienen las células en el estado de mitosis y causan apoptosis. Los compuestos que modulan la actividad de tubulina pueden dividirse de forma general en dos grandes grupos. En primer lugar están los inhibidores de la tubulina como la colchicina y la vincristina, que se unen a ésta impidiendo que forme microtúbulos. Por otro lado, están los agentes estabilizantes de microtúbulos (MSAs), como el paclitaxel y el docetaxel, los cuales se unen preferentemente a la tubulina ensamblada, minimizando la disociación de la tubulina-GDP de los extremos de los microtúbulos e induciendo el ensamblaje de la tubulin-GDP normalmente inactiva. Los fármacos moduladores de la polimerización de microtúbulos han sido muy usados en la terapia antitumoral. Al ser indispensables para la mitosis y detenerla, se logra actuar contra el tumor, pero también se ven afectados aquellos tejidos en continua proliferación (médula ósea, pelo, etcétera). El éxito clínico del paclitaxel y el docetaxel, ha conducido a la búsqueda de nuevos compuestos con el mismo mecanismo de acción y al descubrimiento en los últimos años de una gran cantidad de agentes estabilizantes de microtúbulos con al menos dos sitios de unión distintos.
¿ DESARROLLO DE EPOTILONAS
Como sucedió con paclitaxel, la busqueda por nuevos productos naturales recibió especial atención por diferentes grupos de investigadores. Las epotilonas A y B fueron descubiertas en 1987, por Hofle, Reichenbach y sus colaboradores del centro de investigación Gesellschaft fur Biotechnologische Forschung (GBF), en Alemania. Estos compuestos fueron aislados a partir de extractos de cultivos de degradación de La celulosa por la bacteria Sorangium cellulosum. Por presentar actividad contra del Mucor hiemalis, como las epotilonas A y B fueron primeramente evaluadas como potenciales fungicidas y agrotóxicos. No se utilizaron en tanto algunos experimentos probaban que eran compuestos muy tóxicos. En 1995, un importante descubrimiento se dio gracias a investigadores de Merck en los Estados Unidos que, de manera independiente, aislaron las epotilonas A y B, para comprobar su potente actividad antitumoral con valores IC50 próximos a los del paclitaxel. Otro importante descubrimiento fue que el mecanismo de acción de las epotilonas A y B, no combate al cáncer, era similar al del taxol, exhibiendo potentes propiedades en la estabilización de la tubulina, impidiendo la replicación celular. Las epotilonas A y B mostraron ser capaces de inhibir la glucoproteína P, responsable del desarrollo de la resistencia a los fármacos (Figura 3).
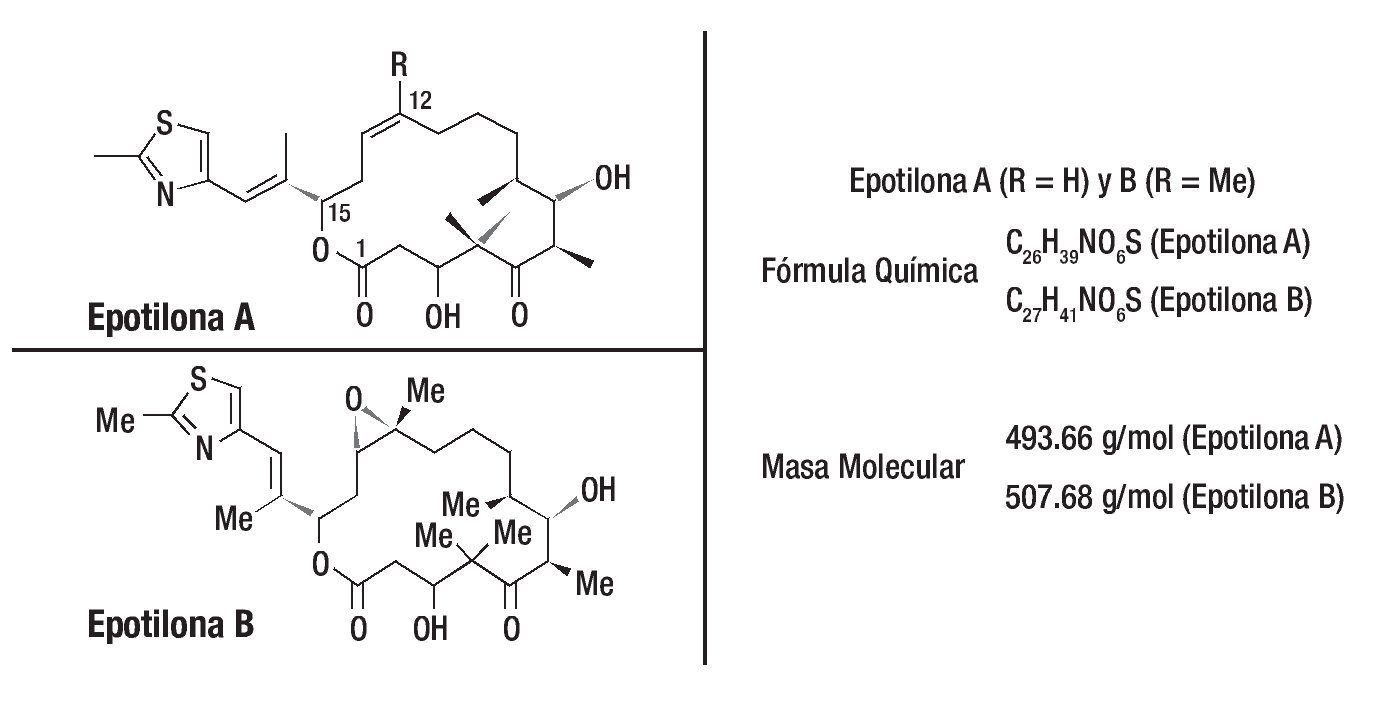
Figura 3. El descubrimiento de las epotilonas fue partir de productos naturales y posteriormente se caracterizaron estas moléculas. Una visión general de las propiedades terapéuticas en estudios preclínicos de los miembros más prometedores de esta clase de drogas se demostró al menos en siete de las epotilonas caracterizadas a través del uso de diferentes modelos de tumores humanos xenotrasplantados. Hasta la fecha, dos productos naturales y análogos semisintéticos han sido evaluados en pacientes con cáncer.
Por esta razón, las epotilonas presentan actividad superior al paclitaxel en células resistentes, siendo que estos resultados fueron, posteriormente, confirmados por científicos del GBF. Debido a los buenos resultados preliminares, la epotilona B pasó a los estudios clínicos de fase I, estando actualmente en fase II. La necesidad de obtener fármacos más potentes, otros análogos fueron sintetizados por varios investigadores, siendo la primera síntesis de epotilona en estado sólido descrita por Nicolau y colaboradores, lo que permitió la obtención de una biblioteca combinada con numerosos análogos. Gracias a los estudios realizados por académicos e industriales, el BMS-247550, un prometedor análogo de la epotilona B, fue producido por la Bristol-Myers Squibb. Este compuesto mostró actividad en varios estadios de estudios clínicos de fase I, estando actualmente en el inicio de la fase II. Estudios de las relaciones entre estructura y actividad biológica realizados por Danishefsky y colaboradores, fortalecieron la epotilona D, conocida también como desoxiepotilona B o KOS-862. Los estudios clínicos de fase I fueron iniciados en octubre de 2001, por la compañia Kosan Bioscience y, actualmente, se encuentra en estudio clínico fase II. La ixabepilona pertenece a una clase de agentes antineoplásicos, que detiene el crecimiento de células tumorales al impedir la multiplicación celular (Figura 4).
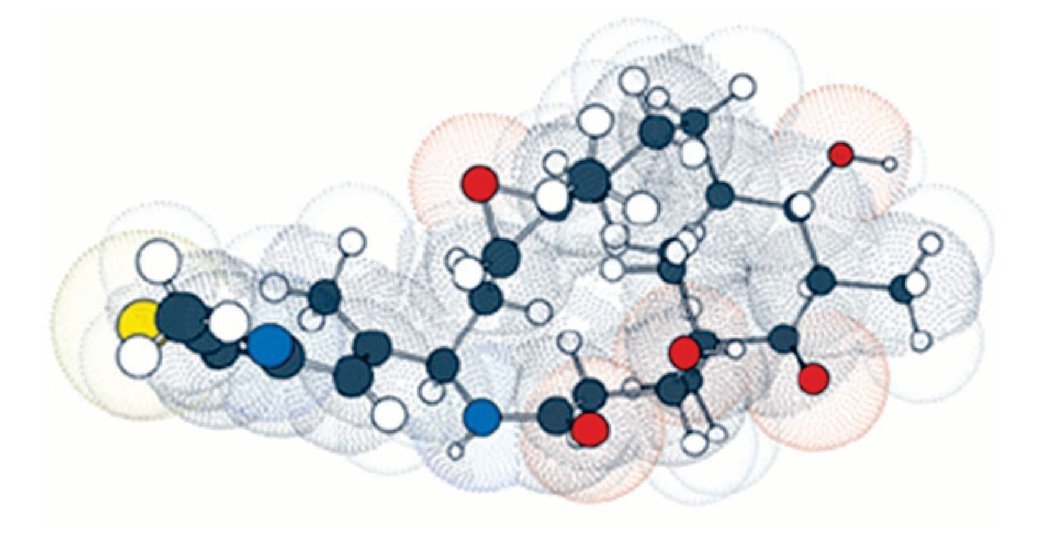
Figura 4. La ixabepilona es un inhibidor de microtúbulos que pertenece a una clase de agentes antineoplásicos llamadas epotilonas. Las epotilonas fueron aisladas del Cellulosum sorangium myxobacterium. La ixabepilona es un análogo semisintético de la epotilona B, un macrólido policétido de 16-carbonos.
Puede utilizarse en forma directa o en combinación con capecitabina para el tratamiento de pacientes resistentes a las antraciclinas y taxanos o cuyo cáncer es resistente a los taxanos y para quienes está contraindicada otra terapia con antraciclinas. La ixabepilona fue aprobada por la Food and Drugs Administration (FDA), en Estados Unidos el 16 de octubre de 2007, y por la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica (ANMAT) en Argentina el 24 de enero de 2008, en base a los resultados beneficiosos y al perfil de seguridad que demostraron los resultados preliminares de los ensayos clínicos desarrollados. La dosis recomendada de ixabepilona es de 40 mg/m2 en forma de infusión intravenosa durante tres horas cada tres semanas. Las dosis para pacientes con un área de superficie corporal (BSA) superior a 2.2 m2 se debe calcular con base en 2.2 m2. La combinación de ixabepilona con capecitabina ha conseguido aumentar en 40% la supervivencia libre de progresión de enfermedad en mujeres con cáncer de mama metastásico que habían progresado rápidamente después de haber recibido quimioterapia. Es la conclusión de un ensayo clínico de fase III presentado en la Conferencia Europea de Cáncer, que se desarrolló en Barcelona. Su uso en combinación con capecitabina, según los resultados de la investigación, prolonga de cuatro a cinco meses el tiempo sin progresión la enfermedad, según señala la citada compañía en un comunicado. BMS espera presentar los primeros datos de supervivencia de este ensayo este 2008 y por el momento no se ha solicitado en la EMEA la autorización para su comercialización aunque, si bien la FDA estadounidense está evaluando el fármaco. El estudio, en el que participaron 752 mujeres de diferentes países con cáncer de mama metastásico previamente tratadas y resistentes a la quimioterapia convencional, es el primer ensayo clínico de fase III que se presenta en Europa con un medicamento perteneciente a la familia de las epotilonas.
¿ DIFERENCIAS ESTRUCTURALES ENTRE EPOTILONAS Y TAXANOS
Los microtúbulos intervienen en diversos procesos celulares que involucran desplazamiento de vesículas de secreción, movimiento de orgánulos, transporte intracelular de sustancias, así como en la división celular (mitosis y meiosis) y que, junto con los microfilamentos y los filamentos intermedios, forman el citoesqueleto. Además, constituyen la estructura interna de los cilios y los flagelos. El sitio de unión de taxol (paclitaxel) en los microtúbulos es una diana farmacológica cuya función es todavía desconocida. Diversas sustancias naturales químicamente diferentes mimetizan los efectos citotóxicos de taxol, aparentemente uniéndose al mismo sitio ubicuo de unión de taxol en los microtúbulos. Entre estas se encuentran las epotilonas (de mixobacterias), la discodermolida (de esponjas marinas) y eleuterobina (de un coral marino). Estos compuestos, llamados agentes estabilizantes de microtúbulos, los estabilizan y bloquean su dinámica; de esta forma detienen la división celular. Por ello taxol se utiliza en la quimioterapia de cáncer de ovario, cáncer de mama metastásico, cáncer de cabeza y cuello y en cáncer de pulmón. El sitio de unión de taxol se ha mapeado en la subunidad de β-tubulina en el lumen de los microtúbulos. Este sitio en la superficie interna de los microtúbulos dificultaría en principio el acceso de los ligandos del sitio de taxol en microtúbulos ensamblados. Hemos estudiado la cinética de unión de un taxoide fluorescente (Flutax-2) a microtúbulos celulares. El sitio de unión es perfectamente accesible a Flutax-2 en citoesqueletos nativos de células PtK2. La cinética de unión y disociación observada es similar a la obtenida con microtúbulos in vitro, conteniendo proteínas asociadas (MAPs). Los datos cinéticos indican que los taxoides se unen directamente de la solución a un sitio expuesto en el microtúbulo. También se ha observado la existencia de un complejo ternario específico de citoesqueletos nativos de PtK2, Hexaflutax y un anticuerpo monoclonal frente a fluoresceína dando lugar a evidencia directa de que el sitio de unión de taxol está expuesto. Por otra parte, también se ha encontrado que cicloestreptina es el primer estabilizador de microtúbulos que se une covalentemente a estos y sus efectos sobre los microtúbulos celulares son irreversibles. La utilización de este ligando ha permitido elucidar el camino que el taxol sigue para alcanzar su sitio en los microtúbulos y además describir un nuevo sitio de unión de agentes estabilizantes en la superficie externa de estos. Esta unión covalente permite que se pueda vencer la resistencia múltiple a drogas de una línea celular que sobre-expresa glicoproteína-P. Los resultados clínicos de la quimioterapia con taxol son a veces insatisfactorios, ya que en muchos casos aparece la llamada resistencia múltiple a drogas (MDR), debido a la sobreexpresión de bombas de membrana, en particular de la glucoproteína P, que expulsa las drogas al exterior celular reduciendo la concentración intracelular de estas. Se trata de ixabepilona la primera de una nueva clase de drogas, conocidas como epotilonas. Esta alternativa terapéutica, desarrollada íntegramente en los centros de investigación de laboratorio, constituye una herramienta fundamental para los oncólogos y las pacientes que generan resistencia o son refractarias a los tratamientos de elección actuales, basados en antraciclinas, taxanos y capecitabina.
¿ PAPEL DE LAS EPOTILONAS EN PACIENTESCON CÁCER DE MAMA RESISTENTES A TAXANOS
Los transportadores multidroga pertenecientes a la ATP-binding cassette (ABC-transporters) son una superfamilia de genes que codifican diferentes proteínas transportadoras (ABCB1, ABCC1, ABCG2, entre otras), responsables de la resistencia a muchos fármacos antineoplásicos. Dichos transportadores generalmente expulsan al medio extracelular los fármacos, aunque pueden inmovilizarlos en el interior de vesículas endocíticas. Algunos tipos de cáncer son muy resistentes a los agentes antineoplásicos, presentando fenotipos de resistencia a múltiples fármacos (MDR). El fenómeno de la resistencia a la quimioterapia es entonces un impedimento para que el tratamiento final del cáncer sea un éxito; aunque se han propuesto diferentes alternativas para vencerla. Uno de los transportadores más estudiados es la glucoproteína P (Pgp), responsable de la resistencia a taxanos, epipodofilotoxinas y antraciclinas, entre otros. En las células neoplásicas, la sobre-expresión de Pgp puede contribuir a la quimiorresistencia en diversos tipos de cáncer y leucemias, explicando la aparición del fenotipo MDR después de una recidiva. La citometría de flujo funcional permite el estudio del fenotipo MDR mediante la utilización de sondas fluorescentes específicas de los diferentes transportadores ABC y en presencia y ausencia de diferentes inhibidores específicos de cada uno de ellos como el verapamilo, la ciclosporina A, valspodar, o la fumitremorgina C. Entre las nuevas estrategias terapéuticas frente a los fenómenos de quimiorresistencia, se están desarrollando nuevos inhibidores de los transportadores multidroga, anticuerpos para bloquear su actividad, e inhibidores de la diferenciación celular. Otros mecanismos de resistencia están relacionados con las topoisomerasas de DNA, con los niveles de la glutatión-sulfonil (GSH), el compuesto tiólico más abundante en los mamíferos o mutaciones de p53. Los fluorocromos como la rodamina, Hoechst 33342, entre otros, permiten estudiar el fenotipo de resistencia, mediante captación, retención y la eliminación de sondas fluorescentes. Si la célula expresa transportadores multidroga, la citometría de flujo permite conocer la cinética del transportador, un dato muy importante porque la incrementada velocidad de expulsión implica mayor quimiorresistencia. Son importantes los diseños experimentales con transfectantes estables de transportadores multidroga (por ejemplo: ABCG2) y en este grupo de investigación del HUVH utilizan células quimiorresistentes a diferentes agentes antineoplásicos como la mitoxantrona o el topotecan. Pero otros aspectos importantes de estos estudios tratan de comprender como los propios agentes anticancerígenos contribuyen a la acumulación de p53 mutante, a la aneuploidización, y a la amplificación de genes implicados en una elevada quimiorresistencia.
Correspondencia: Horacio Astudillo de la Vega M.D., Ph. D.
Unidad de Investigación Médica en Enfermedades Metabólicas, Hospital de Cardiología, Centro Médico Nacional Siglo XXI, IMSS. Avenida Cuauhtémoc N° 330, Colonia de los Doctores. CP 06720, México,
D. F. Teléfono (525) 5627 6900, extensión 22154.
Correo electrónico:hastud2@aol.com