





El carcinoma de células de Merkel (CCM) es una neoplasia neuroendocrina rara de la piel. Fue descrita en 1972 por Toker. Por su origen, expresa marcadores epiteliales y neuroendocrinos, así como falta de características morfológicas distintivas, lo que hace necesario estudio de inmunohistoquímica. Es de curso agresivo, con alto riesgo a recurrencia tanto locorregional como a distancia. El tratamiento definitivo es quirúrgico con márgenes amplios en enfermedad localizada, existe mejoría de control local y de la sobrevida global con radioterapia adyuvante. El uso de quimioterapia esta sólo indicada en enfermedad avanzada o recurrente.
Merkel cell carcinoma (MCC) is a rare malignant neuroendocrine skin cancer, described by Toker in 1972. It express epithelial and neuroendocrine markers by its origin, doesn't have its own morphologic characteristics, that's why immunochemistry has an important role. It has a highly malignant course with a high risk to loco-regional and distance recurrence. Surgery with wide margins is the definitive treatment in localized disease; adjuvant radiotherapy improves in local and overall survival. The chemotherapy is just indicated in an advanced or metastasic disease.
¿ INTRODUCCIÓN
El carcinoma de células de Merkel (CCM) es una neoplasia maligna cutánea rara, descrita por primera vez en 1972 por Toker.1 Su probable origen corresponde a las células de la cresta neural, que generan las células redondas de Merkel, localizadas en el estrato basal de la epidermis y contienen gránulos secretores. Su etiología no está del todo comprendida, existen varios factores de riesgo relacionados con su patogénesis, los cuales incluyen exposición a luz ultravioleta, antecedente de carcinoma de células escamosas o carcinoma de células basales, exposición arsénico, inmunosupresión (pacientes trasplantados, SIDA) o con neoplasias hematológicas. Recientemente, se ha descrito un virus poliomavirus de células de Merkel presente en el 80% de las muestras.2-4 Se han documentado alrededor de 2 000 casos, desde la primera descripción en 1972. La incidencia es de 0.01-0.23 por 100 000 mil habitantes, siendo más común en personas de raza blanca.1 El 78% se presenta en mayores de 59 años.3 Afecta a ambos géneros, siendo más frecuente en hombre.2 Se desarrolla en áreas expuestas al sol, como son cabeza y cuello (50.8%), extremidades superiores e inferiores (35% a 40%) y tronco (10%),3 aunque también puede presentarse en vulva, pene, faringe o mucosas.2 La presentación clínica está dada por una lesión de crecimiento rápido, pétrea, no dolorosa, eritematosa.2
El diagnóstico definitivo es por medio de inmunohistoquímica con citoqueratinas (CK) de bajo peso molecular, predominantemente CK 20, neurofilamento, enolasa neuronal específica (ENE), además de marcadores neuroendocrinos tales como cromogranina, sinaptofisina, péptido intestinal vasoactivo, polipéptido pancreático, calcitonina, sustancia P.1,4,5 Su patogenia no es del todo clara, pero se conocen alteraciones en las vías de señalización, tales como Raf/MEK/ERK, en la cual está involucrada las quinasas fosfatidilinositol 3 quinasa (PI3K) y Akt quinasa. La participación de PTEN, la deleción homóloga de fosfatasa y tensinógeno en cromosoma 10, se considera que tiene un papel relevante en la patogénesis de la enfermedad, esto debido a la frecuencia con la cual se presenta la pérdida heterocigótica del brazo largo del cromosoma 10 (LOH).6-8 La existencia de un poliomavirus de células de Merkel (MCV), descrito en 1953, podría explicar oncogénesis viral. Éste ADN virus interacciona con las células del hospedero mediante un antígeno T, a través de gen p53 y retinoblastoma (RB), con esto influirían en el ciclo celular de la célula huésped. Se ha encontrado hasta un 80% de integración del virus a las células neoplásicas. Su impacto clínico se está dilucidando, un estudio recién publicado de Antoine Touzé, Emmanuelle Le Bidre y colaboradores de 67 pacientes, demostraron la presencia de MCV en el 100% de los pacientes, de los cuales el 64.7% tuvieron títulos de anticuerpos por arriba de 10 000, quienes se asociaron a un periodo libre de progresión mayor (Hazard Ratio 4.6; 95% IC 1.7 a 12.2; p=0.002).9
¿ PRESENTACIÓN DEL CASO
Paciente masculino de 70 años, costurero, inicia en junio del 2010 con lesión en tercio medio lateral de pierna izquierda de 2 cm, con crecimiento progresivo y posteriormente nueva lesión en región inguinal (Figuras 1A y 1B). Por este motivo fue valorado en su hospital de zona, donde se le realiza biopsia con reporte de neoplasia maligna, siendo referido a nuestro Hospital para complementación diagnóstica y terapéutica, se realizó revisión de material histopatológico:
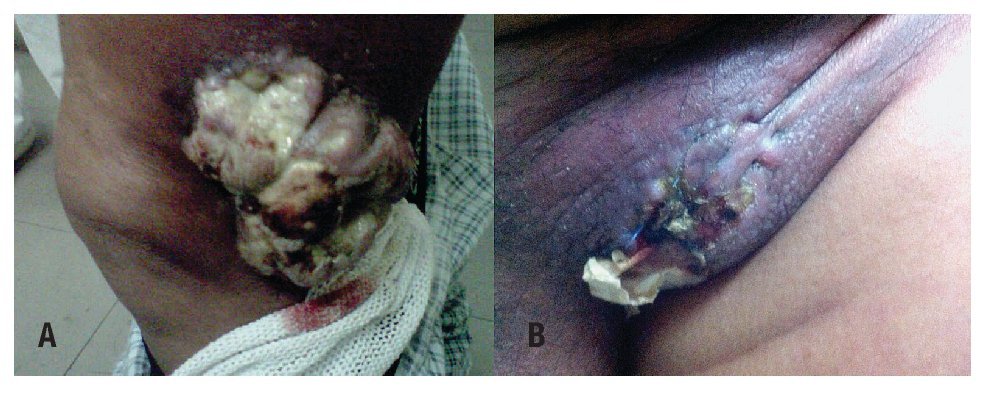
Figura 1. Lesión de tejidos blandos ulcerada, fungada, voluminosa, lateral a rodilla izquierda A). Lesión ulcerada región inguinal izquierda B).
Hallazgos patológicos: Microscópicamente, se observa neoplasia que crece por debajo de la epidermis e infiltra en forma difusa la dermis, extendiéndose hasta el tejido celular subcutáneo. Infiltra en forma difuso con patrones de crecimiento trabecular, y en mantos sólidos de tamaño variable. La epidermis se encuentra respetada (Figuras 2A y 2B). Las células neoplásicas son redondas de tamaño homogéneo y apariencia monótona. Presentan citoplasma escaso, núcleos redondos, con cromatina granular y múltiples nucleólos. Además, se identifican numerosas mitosis y cuerpos apoptóticos (Figura 2C). El estroma presenta septos fibrosis con numerosos vasos capilares. Inmunohistoquímica: Por métodos de inmunohistoquímica, las células neoplásicas mostraron positividad para cromogranina, sinaptofisina y CD56. Las células neoplásicas también mostraron intensa positividad para Ki-67, con un índice de proliferación celular del 70%. Además, presentaron intensa expresión para p53 y negativas para p63, concluyendo con el diagnóstico de Carcinoma de Células de Merkel.
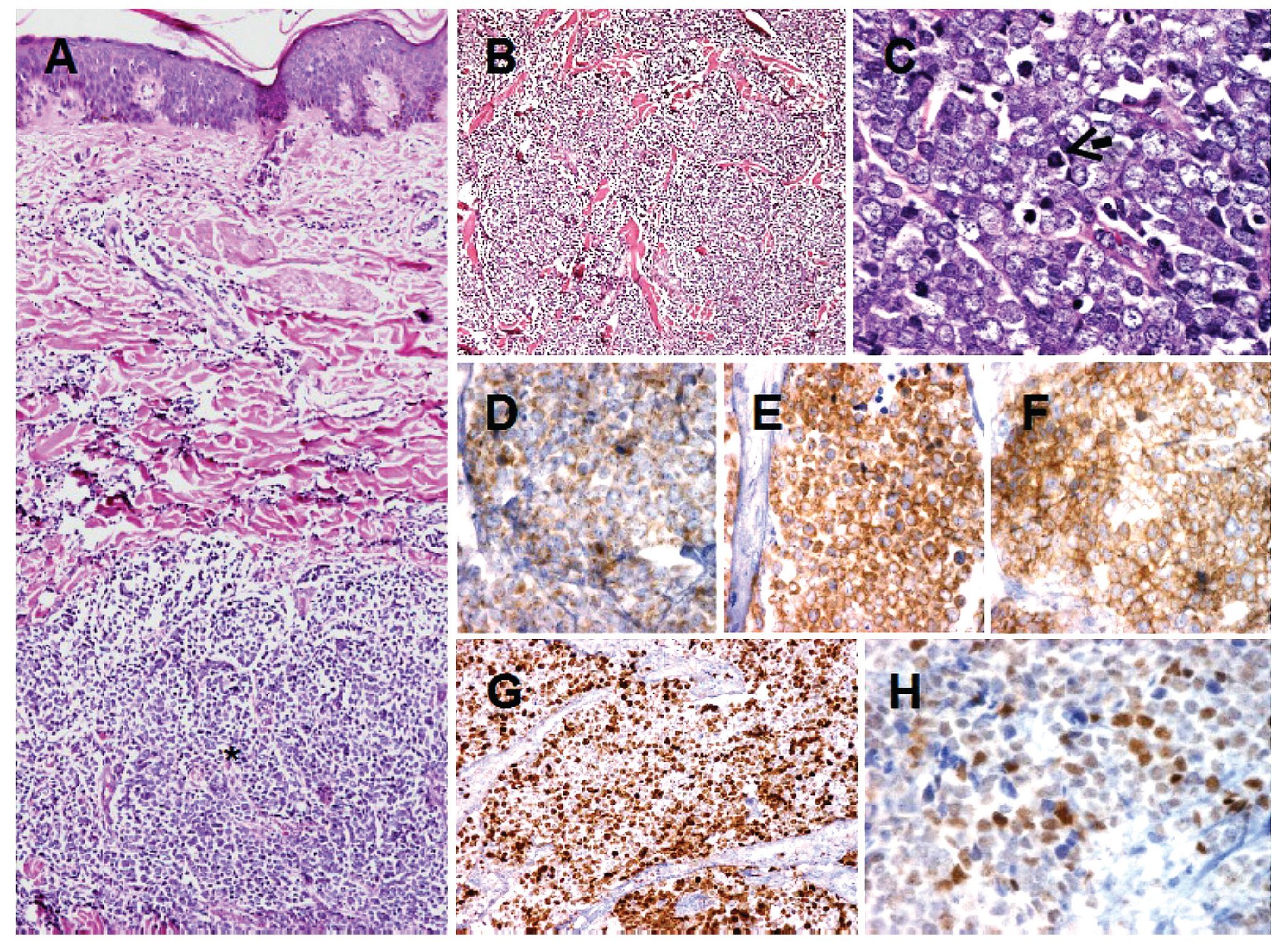
Figura 2. Carcinoma de células de Merkel. Se observa neoplasia que crece en la dermis (asterisco) y que respeta la epidermis A). El tumor presenta crecimiento difuso con patrón trabecular. Las células neoplásicas son redondas de tamaño homogéneo y apariencia monótona B). Las células neoplásicas muestran cromatina finamente granular y numerosas mitosis (flecha) C). Por inmunohistoquímica, las células neoplásicas muestran positividad citoplasmática para cromogranina D). Células positivas para sinaptofisina E). Células positivas para CD56 F). Intensa positividad para Ki-67 G). Las células muestra expresión para p53 H).
Se realizó tomografía computada de tórax y abdominopélvica que evidenció actividad tumoral retroperitoneal (Figura 3), retrorrenal izquierda de 5.5 cm, infiltración pélvica con extensión a cavidad pélvica de 12.2 x 11.5 cm, infiltración inguinal izquierda de 7.3 x 10.8 cm, infiltración de tejido dérmico subcutáneo en tercio medio de miembro pélvico izquierdo en cara lateral, sin afectar capa muscular.
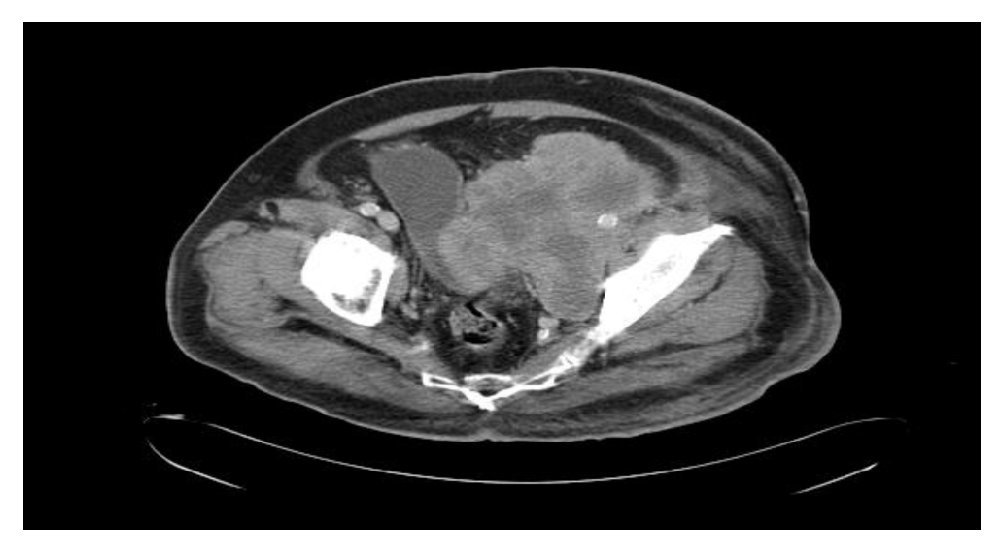
Figura 3. Tomografía con actividad tumoral en retroperitoneo.
Valorado por Servicio de Radioterapia, quienes consideraron candidato a recibir radioterapia a 60Gy en 30 fracciones, las cuales recibe actualmente. Fue enviada a Servicio de Oncología Médica para inicio de tratamiento sistémico, cuenta con ECOG 1, no siendo candidato a tratamiento concomitante, por no existir beneficio en sobrevida global. Lo consideramos candidato para manejo secuencial, con prioridad de radioterapia paliativa para el dolor y la hemorragia, secundaria a la neoplasia.
¿ DISCUSIÓN
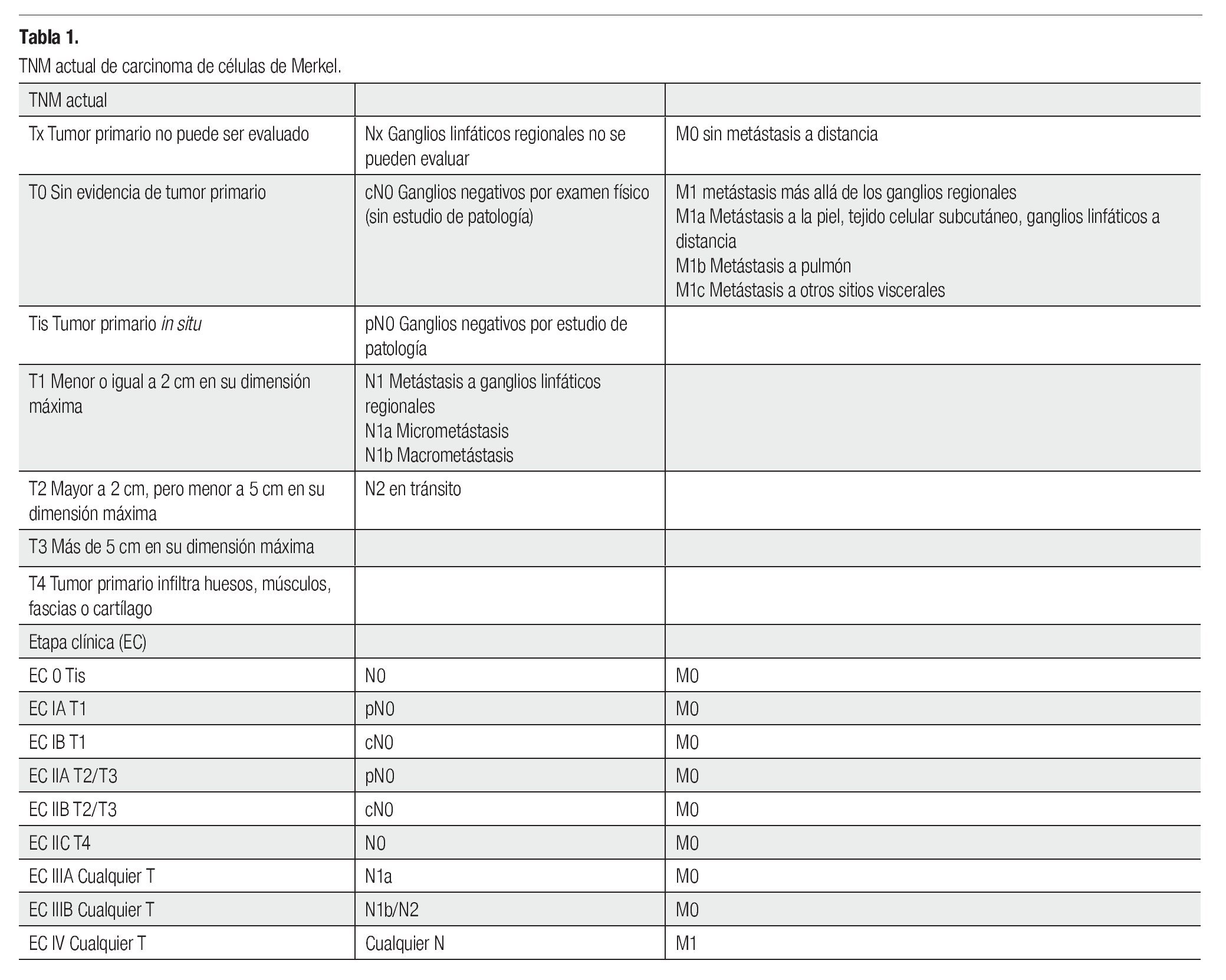
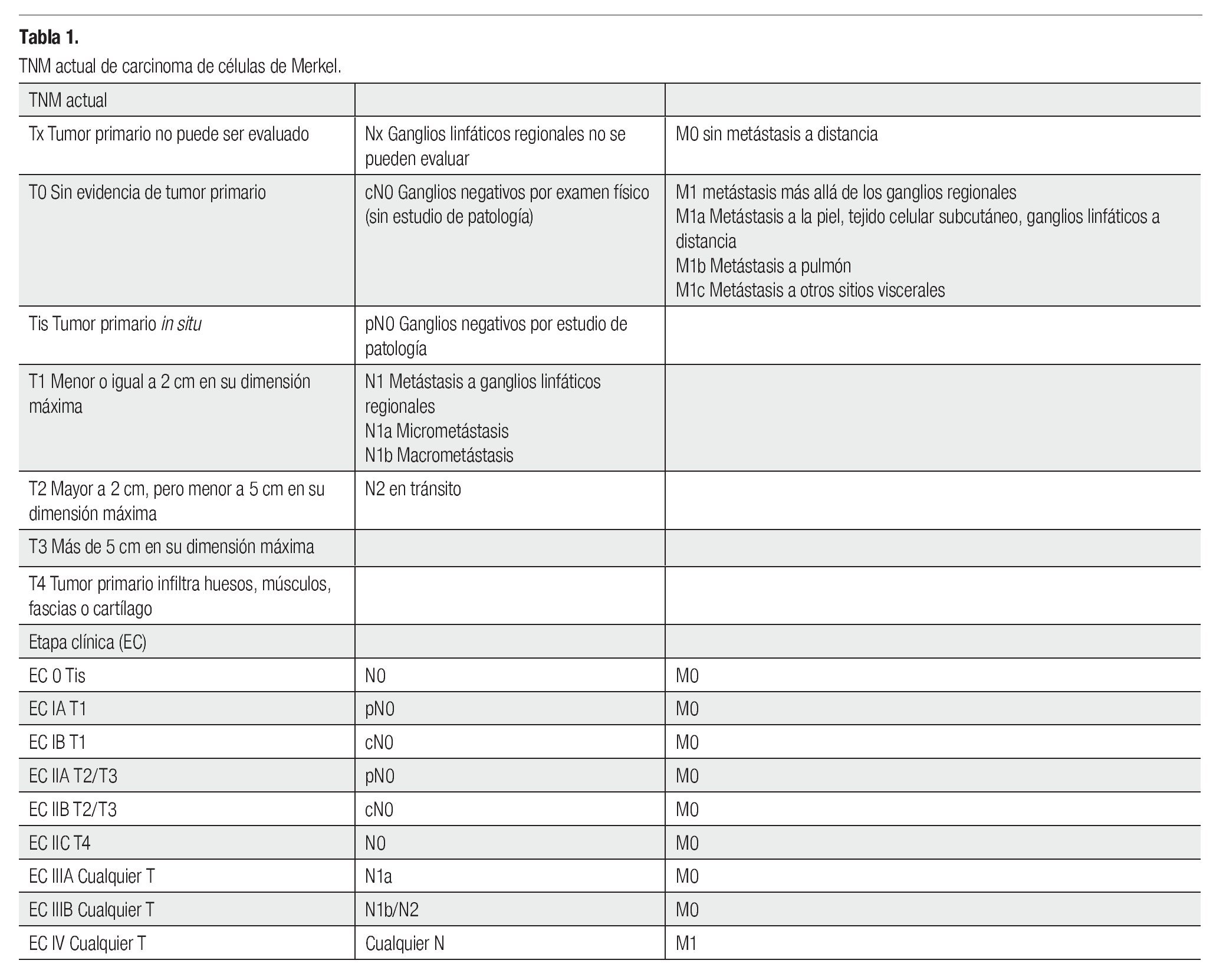
El CCM consiste en una entidad rara, con pobre pronóstico (sobrevida a cinco años del 0% al 68%). Los factores que afectan a la sobrevida son el estado ganglionar, enfermedad a distancia, recurrencia y tamaño del tumor, así como márgenes estrechos en la cirugía.1-5,10-13 La etapificación ha sufrido cambios constantes desde la clasificación de 1992, hasta la actual del 2010 (Tabla 1).1-3
MANEJO DEL CCM
Cirugía: Es la piedra angular del CCM, siempre y cuando sea posible (etapa clínica I, II, N1). Dentro de su abordaje están la extensión de márgenes, biopsia del ganglio centinela, así como disección ganglionar.3 Se recomienda márgenes de 3 cm y profundidad cuando sea posible de 2 cm, en base a los estudios de Yiengruksawan y Ottl.1,14 La cirugía de Mohs seguida de radioterapia es usada en lesiones pequeñas, reduciendo metástasis persistentes, en tránsito y enfermedad nodal.3,14,15 La biopsia de ganglio centinela se recomienda en lesiones iguales o mayores de 1 cm, por el riesgo a metástasis nodal según reportes de estudios de Clark JR, Stawowy y Allen, y con disección ganglionar en caso de positividad. Sino es posible, se debe otorgar radioterapia adyuvante tanto al primario, como a la región nodal.3,5,15-17
Radioterapia: Es una neoplasia radiosensible, con reportes como los del Peter MacCallum Cancer Institute, donde han documentado respuesta completa del tumor medible del 96%, y 4% de respuesta parcial (tasa respuesta global del 100%).17-21 La radioterapia adyuvante ha mostrado mejorar recurrencia local y sobrevida media. Un estudio del MD Anderson Cancer Center, mostró que la radioterapia disminuyó riesgo de recurrencia local (p<0.00001).20,21 Otro estudio evidenció beneficio en sobrevida media, a pacientes que recibieron radioterapia adyuvante 63 meses vs 45 meses quienes no recibieron, especialmente a tumores de más de 2 cm. Un estudio australiano de 72 pacientes, demostró que los pacientes que sólo eran sometidos a cirugía (38 pacientes) recaían con tiempo medio de recaída (TMR) de 5.5 meses vs el grupo que recibió radioterapia adyuvante, donde sólo recayeron 10 de 34 con TMR de 16.5 meses.3-5
Quimioterapia: El estudio TROG 9607 evaluó el tratamiento con radioterapia al sitio principal de enfermedad y a los nódulos, en tratamiento concomitante con carboplatino a un AUC de 4.5 y etopósido a dosis de 80 mg/m2, encontrando mejoría del control locorregional y sobrevida con la aplicación de quimioterapia. No obstante, en otros tres estudios posteriores no reprodujeron dicho beneficio.3,22 El manejo del CCM metastásico con quimioterapia da tasas de respuesta de hasta 70%, pero la duración de ésta es corta y su impacto en sobrevida global es poca o casi nula. El tratamiento es multimodal, ya sea con radioterapia o cirugía. A pesar de que existen varios esquemas de quimioterapia simples o en combinación, los cuales incluyen terapias de combinación de cisplatino con etopósido, doxorrubicina, vincristina con ciclofosfamida, o mitoxantrona, epirrubicina, bleomicina, ifosfamida, no existe un esquema estándar de tratamiento. La sobrevida global es de 10 meses en enfermedad metastásica.1,4,5,23-28 Hay reportes anecdóticos de cura, al utilizar factor de necrosis tumoral e interferón.2 Se están evaluando inhibidores de mTOR, cuyos reportes están aún en espera.3-5
Correspondencia: Dr. Miguel Quintana Quintana Médico.
Av. Cuauhtémoc 330, Col. Doctores, Del. Cuauhtémoc. C.P. 06720. México D.F., México.
Teléfono: 55 5627 6900.
Correo electrónico:qquintanam@gmail.com