









La metagenómica ha impulsado de manera decisiva el estudio del microbioma intestinal, lo que ha permitido comprender su importancia para la salud humana. La metataxonómica, basada en la secuenciación del gen del ARNr 16S, ofrece perfiles taxonómicos de procariotas, mientras que la metagenómica shotgun permite una caracterización más completa de todo el ADN presente en la muestra. Con una profundidad de secuenciación adecuada, esta última amplía la resolución taxonómica hasta el nivel de cepa y proporciona información detallada sobre el potencial funcional de la microbiota. Sin embargo, la falta de estandarización en la recolección y procesamiento de muestras, las tecnologías de secuenciación, y la interpretación y gestión de los datos, limita la comparación de resultados y su implementación en laboratorios clínicos. Esta revisión ofrece un marco práctico y actualizado sobre las metodologías metagenómicas, el análisis de datos y el uso de inteligencia artificial, destacando avances y buenas prácticas para facilitar su integración en la práctica médica y superar los desafíos actuales.
Metagenomics has decisively advanced the study of the gut microbiome, enabling a better understanding of its importance for human health. Metataxonomics, based on the sequencing of the 16S rRNA gene, provides taxonomic profiles of prokaryotes, while shotgun metagenomics allows a comprehensive characterization of all DNA present in a sample. With adequate sequencing depth, the latter increases taxonomic resolution to the strain level and provides detailed information on the functional potential of the microbiota. However, the lack of standardization in sample collection and processing, sequencing technologies, and data management limits the comparability of results and their implementation in clinical laboratories. This review offers a practical and updated framework on metagenomic methodologies, data analysis, and the application of artificial intelligence tools, highlighting advances and best practices to facilitate the integration of functional microbiome analysis into clinical practice and to overcome current challenges.
La microbiota humana es el conjunto de microorganismos, incluyendo bacterias, virus, protozoos y hongos, que cohabitan en simbiosis en diferentes localizaciones del cuerpo humano, con grupos de especies estables y otras variables. De los distintos microambientes específicos del cuerpo, el tracto gastrointestinal es el más investigado, en concreto la microbiota intestinal, por su facilidad de estudio a través de las heces. El concepto de microbioma es más amplio e incluye la microbiota, sus genomas y metabolitos, y las condiciones del ambiente circundante.
Históricamente, los estudios basados en técnicas de cultivo han contribuido a generar información básica sobre los microorganismos, especialmente aquellos relacionados con la patología infecciosa. Sin embargo, hoy sabemos que la mayoría de los microorganismos intestinales no son cultivables en los medios de cultivo y condiciones tradicionales1. El conocimiento del microbioma intestinal se ha impulsado muy notablemente en los últimos años gracias a la aplicación de las técnicas de secuenciación masiva o NGS (next-generation sequencing)1. Por otro lado, estos estudios masivos del microbioma han contribuido a comprender que, más que la composición microbiana, la importancia del microbioma en relación con la salud humana radica en su funcionalidad2. Se estima que el microbioma intestinal contiene un número de genes muy superior al del genoma humano3, donde diferentes especies microbianas pueden llevar a cabo funciones metabólicas equivalentes y, a su vez, una misma especie puede realizar diversas funciones. Por tanto, es evidente que existe una necesidad de analizar las propiedades funcionales del microbioma, y la relación de éstas con las del propio individuo. Todo ello pone de manifiesto la necesidad de avanzar en el campo de la Biología de Sistemas, tanto para facilitar la integración de metadatos procedentes de diversas fuentes (por ejemplo, aquellos relacionados con el hospedador), como para optimizar el diseño de modelos metabólicos que permitan analizar dicha información en un contexto biológicamente significativo. En los últimos años, se han desarrollado nuevos métodos analíticos y herramientas computacionales que permiten analizar de forma global el conjunto de datos relacionados con las características biológicas del microbioma humano. Estos datos derivan de diferentes aproximaciones ómicas, que incluyen la culturómica (empleo de múltiples condiciones de cultivo combinado con herramientas avanzadas de identificación microbiana), la metataxonómica (estudio de la composición y cantidad relativa de microorganismos), la metagenómica (estudio de la composición y funcionalidad de la microbiota), la metatranscriptómica (estudio de la expresión génica de la microbiota), la metaproteómica (estudio de la síntesis de proteínas por parte de la microbiota) y la metabolómica (estudio de los metabolitos originados por la microbiota), entre otras (fig. 1). La recolección de todos estos datos y, especialmente, su posterior análisis, es una labor compleja y costosa, que actualmente sólo es posible llevar a cabo en algunos laboratorios de investigación. Por el momento, no se ha podido incorporar a la mayoría de los laboratorios asistenciales ya que para ello se necesitaría estandarizar los procesos y protocolos y, sobre todo, establecer rangos de normalidad, además de incorporar personal bioinformático en los hospitales. Estos enfoques, aunque laboriosos, están ampliando significativamente nuestro conocimiento del microbioma intestinal y sus posibles aplicaciones preventivas y terapéuticas, pero hoy en día aún no son una realidad en los hospitales asistenciales4.
 siembra de los microorganismos en múltiples medios y protocolos de cultivo adecuados y aislamiento masivo de microorganismos (culturómica); 2) extracción y secuenciación del ADN microbiano (metataxonómica y metagenómica); 3) extracción y secuenciación del ARNm microbiano (metatranscriptómica), y 4) extracción y análisis de los metabolitos microbianos (metabolómica). Estas técnicas requieren herramientas computacionales avanzadas y laboratorios especializados, dado el volumen y la complejidad de los datos generados. Elaboración propia a partir de recursos gráficos disponibles en BioRender.")
Aproximaciones metodológicas para el estudio del microbioma intestinal humano. A partir de la microbiota intestinal procedente de muestras gastrointestinales/fecales se pueden realizar diversos análisis con distintos objetivos: 1) siembra de los microorganismos en múltiples medios y protocolos de cultivo adecuados y aislamiento masivo de microorganismos (culturómica); 2) extracción y secuenciación del ADN microbiano (metataxonómica y metagenómica); 3) extracción y secuenciación del ARNm microbiano (metatranscriptómica), y 4) extracción y análisis de los metabolitos microbianos (metabolómica). Estas técnicas requieren herramientas computacionales avanzadas y laboratorios especializados, dado el volumen y la complejidad de los datos generados.
Elaboración propia a partir de recursos gráficos disponibles en BioRender.
Una gran limitación para el desarrollo de estos estudios es la estandarización en la recogida de muestras y la extracción de material genético. Ambos factores son fundamentales para garantizar la comparación de resultados y la reproducibilidad interlaboratorios de los estudios metagenómicos. Sin embargo, existen diversos desafíos asociados a esta etapa crítica del análisis. La calidad y composición del microbioma extraído pueden variar considerablemente debido a factores como el tipo de muestra (fecal, tejido de mucosa, uso de hisopos fecales…), el método de almacenamiento, el tiempo transcurrido entre la recogida y el procesamiento, y las técnicas utilizadas para la extracción de ADN. Por ejemplo, estudios han demostrado que la congelación a –80°C y el uso de estabilizadores de ADN, como los presentes en algunos de los tubos de recogida fecal comerciales, son esenciales para minimizar la degradación del material genético y la proliferación bacteriana indeseada5. Si bien existen discrepancias sobre el uso de estos estabilizadores, pues pueden afectar a la determinación de otras moléculas en las muestras de heces como metabolitos6,7. Por otro lado, la eficiencia de los kits de extracción de ADN puede diferir significativamente en función de su capacidad para lisar células y liberar ADN de alta calidad, afectando tanto al rendimiento de la extracción como a la representatividad taxonómica del análisis posterior8. A la hora de estandarizar el proceso se necesita además contemplar su automatización, para facilitar su implantación en un laboratorio clínico evitando así procesos laboriosos y dependientes de la persona que los realiza.
Adicionalmente, la falta de protocolos estandarizados introduce sesgos en el perfilado del microbioma, ya que las diferencias en las técnicas de extracción y almacenamiento pueden resultar en la pérdida de taxones específicos, particularmente los infrarrepresentados, o en la sobreestimación de los más abundantes. Estos sesgos, a su vez, limitan la posibilidad de realizar comparaciones fiables entre estudios o laboratorios9. La variabilidad técnica también afecta a la reproducibilidad de los resultados, particularmente en estudios longitudinales o multicéntricos. Por tanto, establecer protocolos consensuados estándar para la recogida, almacenamiento y procesamiento de muestras se considera un paso clave para avanzar hacia un análisis metagenómico científico más robusto y comparable.
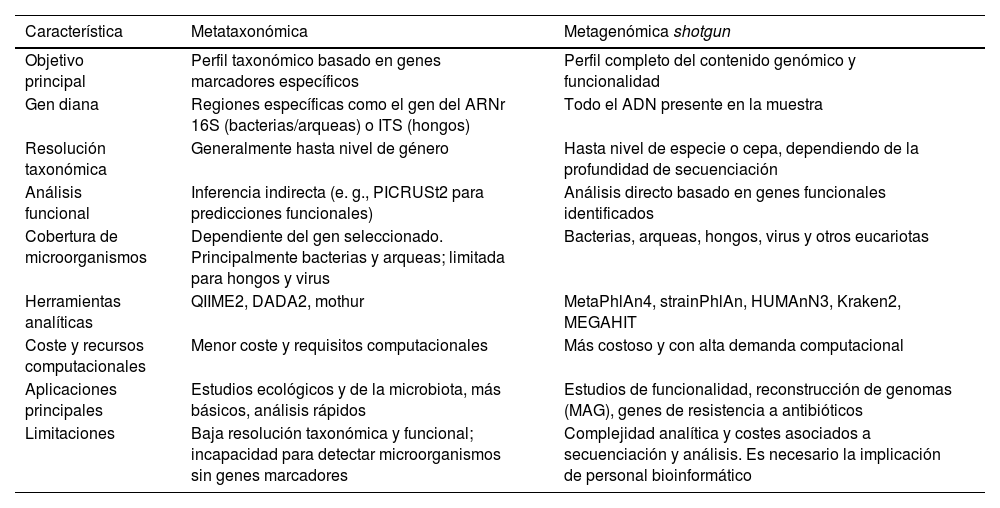
El auge de la metataxonómica o metagenómica filogenéticaLos enfoques basados en la secuenciación del ADN extraído de comunidades microbianas complejas han supuesto un cambio de paradigma en el estudio del microbioma intestinal. Inicialmente, el perfil taxonómico de las comunidades microbianas se obtenía mediante la secuenciación de amplicones de uno o más genes considerados como marcadores filogenéticos (metataxonómica), como el gen que codifica la subunidad 16S del ribosoma bacteriano (gen del ARNr 16S) para procariotas y el gen que codifica la subunidad 18S en el caso de los microorganismos eucariotas (gen del ARNr 18S). Además, en el caso de los eucariotas, se suelen usar las regiones intergénicas espaciadoras transcritas (ITS), situadas entre las subunidades grande y pequeña de los genes ribosomales, como marcadores para diferenciar especies con mayor resolución. La amplificación y la secuenciación de estas regiones permite crear un perfil taxonómico de la comunidad. Para ello, las lecturas de secuenciación se someten a un conjunto de pasos de filtrado con el objetivo de reducir los artefactos técnicos, entre los que se incluye la agregación de lecturas casi idénticas. Esta agregación se puede realizar agrupando secuencias con un valor de similitud determinado (95-99% de identidad), generando una unidad taxonómica operativa (operational taxonomic unit [OTU]), o mediante la creación de variantes de secuencia de amplicón (amplicon sequence variant [ASV]), que ofrecen un nivel de resolución más alto10. Posteriormente, herramientas y programas bioinformáticos como DADA211 o QIIME212 permiten la asignación taxonómica de las secuencias comparándolas con las depositadas en bases de datos como SILVA13, Greengenes214 o RefSeq15. Aunque la secuenciación de amplicones solo permite conocer la información taxonómica de la comunidad microbiana, actualmente existen herramientas bioinformáticas que permiten realizar inferencias funcionales, como PICRUSt216 o Tax4Fun217, que predicen la composición funcional de una comunidad microbiana utilizando una base de datos de genomas de referencia.
La secuenciación de amplicones del gen del ARNr 16S es la metodología más comúnmente utilizada en los estudios de microbiota. No obstante, presenta limitaciones importantes: no permite la detección de virus ni de microorganismos eucariotas, ya que estos carecen de dicho gen, y tampoco proporciona información directa sobre el contenido genómico real. Además, el número de operones ribosomales varía entre especies e incluso entre cepas, lo que impide estimar con precisión la abundancia relativa de un microorganismo basándose únicamente en el número de lecturas del gen del ARNr 16S. Estas limitaciones junto con el hecho de proporcionar únicamente información del perfil composicional han impulsado el desarrollo de la secuenciación a gran escala (tabla 1). La selección del enfoque depende de los objetivos del estudio, el presupuesto disponible y la infraestructura analítica. En general, la metataxonómica puede ser más adecuada para estudios exploratorios, mientras que la metagenómica shotgun se prefiere para estudios integrados o funcionales.
Comparación y principales diferencias entre enfoques metagenómicos
| Característica | Metataxonómica | Metagenómica shotgun |
|---|---|---|
| Objetivo principal | Perfil taxonómico basado en genes marcadores específicos | Perfil completo del contenido genómico y funcionalidad |
| Gen diana | Regiones específicas como el gen del ARNr 16S (bacterias/arqueas) o ITS (hongos) | Todo el ADN presente en la muestra |
| Resolución taxonómica | Generalmente hasta nivel de género | Hasta nivel de especie o cepa, dependiendo de la profundidad de secuenciación |
| Análisis funcional | Inferencia indirecta (e. g., PICRUSt2 para predicciones funcionales) | Análisis directo basado en genes funcionales identificados |
| Cobertura de microorganismos | Dependiente del gen seleccionado. Principalmente bacterias y arqueas; limitada para hongos y virus | Bacterias, arqueas, hongos, virus y otros eucariotas |
| Herramientas analíticas | QIIME2, DADA2, mothur | MetaPhlAn4, strainPhlAn, HUMAnN3, Kraken2, MEGAHIT |
| Coste y recursos computacionales | Menor coste y requisitos computacionales | Más costoso y con alta demanda computacional |
| Aplicaciones principales | Estudios ecológicos y de la microbiota, más básicos, análisis rápidos | Estudios de funcionalidad, reconstrucción de genomas (MAG), genes de resistencia a antibióticos |
| Limitaciones | Baja resolución taxonómica y funcional; incapacidad para detectar microorganismos sin genes marcadores | Complejidad analítica y costes asociados a secuenciación y análisis. Es necesario la implicación de personal bioinformático |
MAG: metagenome-assembled genomes.
La secuenciación shotgun de metagenomas se ha convertido en el método de elección para estudiar y clasificar microorganismos de diversos ecosistemas. La mejora constante de la calidad y la rentabilidad, especialmente el abaratamiento de los costes hace que sea una técnica cada vez más fácil, rápida y asequible en coste y manipulación.
El objetivo de la metagenómica mediante shotgun es la secuenciación no dirigida de todos los genomas presentes en una muestra, lo que permite un perfilado de mayor resolución y el estudio del contenido génico y el perfil funcional18. Este análisis puede realizarse desde dos enfoques distintos.
Por un lado, el ensamblaje de novo trata de reconstruir los genomas a partir de fragmentos de ADN19. Para ello, existen herramientas que agrupan las secuencias en unidades de mayor tamaño o contigs, como SPAdes20 o MEGAHIT21. Estos ensamblados se comparan en bases de datos de referencia y permiten realizar la anotación funcional y taxonómica. No obstante, los contigs también pueden agruparse en conjuntos procedentes del mismo organismo mediante métodos de agrupamiento o binning, y posteriormente, reconstruir genomas ensamblados a partir de los metagenomas (metagenome-assembled genomes [MAG])22. Este abordaje permite analizar en detalle las funciones y el metabolismo de microorganismos y profundizar en sus complejas interacciones y además obtener genomas de microorganismos desconocidos, pero solo si presentan una cobertura adecuada como para ser ensamblados. Sin embargo, requiere un elevado coste computacional como consecuencia del ensamblado, mapeado y binning18. Además, el ensamblaje metagenómico presenta varios retos y no es universalmente aplicable23, por ejemplo, no es adecuado para genomas de baja abundancia o en presencia de diferentes cepas dentro de la misma especie bacteriana.
El segundo enfoque computacional permite identificar la composición taxonómica y el perfil funcional de las lecturas de secuenciación mediante su mapeo en bases de datos de genomas microbianos de referencia o de familias de proteínas19. Para esta aproximación, suelen utilizarse herramientas como MetaPhlAn424, que permite determinar la composición microbiana a nivel de especie, y HUMAnN325, que realiza el análisis funcional. Estos métodos permiten mitigar los problemas de ensamblaje, aumentar la velocidad de computación y detectar microorganismos que presentan una baja abundancia18.
La mayoría de los métodos utilizados para el perfilado del microbioma intestinal están limitados a nivel de especie. Sin embargo, existe una variación considerable dentro de una misma especie, por lo que el análisis a nivel de cepa está ganando mucho interés. En la actualidad, se han desarrollado herramientas que permiten detectar las cepas presentes en una muestra mediante metagenómica shotgun. Un ejemplo es StrainPhlAn26, que identifica la cepa dominante de una especie determinada en cada muestra. Todo ello, de nuevo, es muy dependiente de que todas las secuencias sean depositadas en bases de datos públicas, y sea posible de ese modo identificar cepas específicas que estén diseminadas en varios países, o se asocien a una patología concreta. Por ejemplo, la metagenómica shotgun ha permitido identificar las cepas Escherichia coli (E. coli) O157:H7 y Klebsiella pneumoniae UCI 34 asociadas a pacientes con infección recurrente por Clostridioides difficile27, o el clon ST131 de E. coli, que es el principal causante de las infecciones urinarias en todo el mundo y se asocia específicamente con resistencia a muchos de los antimicrobianos que se usan en esa patología28.
Por otro lado, el viroma es un componente importante de las comunidades microbianas, lo que hace que su caracterización sea fundamental para comprender este ecosistema. Aunque la identificación de virus a partir de metagenomas se lleva realizando desde hace tiempo, las estrategias anteriores requerían el ensamblaje de las secuencias en contigs y la posterior identificación de las secuencias virales dentro de los mismos. En los últimos años, han surgido bases de datos mejoradas de virus intestinales, como el catálogo metagenómico de virus intestinales (Metagenomic Gut Virus cataloge [MGV])29, que ha facilitado el desarrollo de herramientas capaces de perfilar su contenido, como Phanta30. También existen numerosos desafíos en la caracterización del micobioma intestinal. Aunque existen herramientas específicas, como FunOMIC31, MiCoP32 o HumanMycobiomeScan33, actualmente su robustez es limitada, en gran parte debido a la falta de bases de datos de referencia completas y actualizadas34.
Hasta la fecha, los estudios metagenómicos se han realizado principalmente utilizando sistemas de secuenciación de segunda generación, como Illumina o Ion Torrent, que solo permiten la secuenciación de fragmentos cortos de ADN, generalmente de unos 300 pares de bases. La secuenciación de fragmentos largos de ADN presenta ciertas ventajas, especialmente para el ensamblaje de las secuencias. En este sentido, las tecnologías de secuenciación de tercera generación, como PacBio y Oxford Nanopore, han mejorado significativamente su precisión y disminuido su coste, por lo que las está posicionando como herramientas cada vez más utilizadas en el estudio del microbioma intestinal35. Ambas tecnologías se caracterizan por leer fragmentos largos de ADN, pudiendo llegar a tener el cromosoma bacteriano en 2 o 2 contigs. La combinación de ambas tecnologías se está aplicando ya en los laboratorios clínicos para la identificación y análisis de patógenos ocasionales, generalmente poco frecuentes, como fue en su momento el virus del SARS-CoV-236. La estrategia se basa en combinar una tecnología de lecturas cortas, con pocos errores de secuenciación, con otra tecnología que proporcione lecturas largas, aunque esta pueda contener más fallos. Muchos laboratorios asistenciales incorporaron plataformas de secuenciación con la pandemia del COVID-19 que ahora se están usando para otros microorganismos, y que sin duda serán de gran ayuda para a avanzar en la lucha contra las resistencias a los antibióticos gracias a la detección de los mecanismos genéticos implicados.
El análisis de los datos metagenómicos se realiza habitualmente mediante herramientas basadas en línea de comandos, lo que implica un alto nivel de conocimientos en bioinformática. Esto se debe, entre otros motivos, a que se suele trabajar con un elevado número de archivos, lo que hace recomendable automatizar tareas repetitivas mediante scripts; a que, en muchos casos, se requiere una elevada capacidad de computación, normalmente accesible a través de servidores que se manejan por línea de comandos; y a que este entorno permite una mayor personalización de los análisis y el uso de herramientas avanzadas37. No obstante, se han desarrollado plataformas con interfaz gráfica, como MicrobiomeAnalyst (https://www.microbiomeanalyst.ca/), que simplifican significativamente el proceso. Estas herramientas hacen el análisis más accesible para usuarios con conocimientos bioinformáticos limitados, si bien su capacidad de personalización es reducida, lo que puede limitar su utilidad en estudios más complejos o específicos.
Por otro lado, con un enfoque complementario para abordar los desafíos técnicos en el análisis del microbioma, también se ha propuesto el uso de la secuenciación metagenómica shotgun poco profunda (shallow shotgun sequencing [SS]), como una alternativa eficiente a los métodos tradicionales como la secuenciación de amplicones del gen del ARNr 16S y la secuenciación metagenómica profunda38. Esta técnica que utiliza profundidades de secuenciación entre 2 y 5 millones de lecturas por muestra, presenta menos variabilidad técnica que la secuenciación del gen del ARNr 16S en diferentes etapas experimentales, como la preparación de las librerías y muestra una resolución mucho mayor, superando las limitaciones del gen del ARNr 16S para clasificar únicamente a nivel de género en la mayoría de los casos, y permitiendo a su vez una caracterización funcional directa del microbioma mediante el perfilado de genes específicos38.
Limitaciones y retos en la aplicación de la metataxonómica y la metagenómica en la práctica clínicaA pesar de los importantes avances descritos en la sección anterior, el análisis metagenómico continúa enfrentando múltiples desafíos (tabla 2). Por un lado, la calidad de los datos y el perfilado del microbioma están condicionados por factores experimentales, biológicos y ambientales. Entre los más relevantes, destacan el tipo de muestra recogida y el procedimiento o kit empleado, el método de conservación y la técnica y plataforma de secuenciación empleadas. Por ejemplo, se han descrito diferencias en la composición microbiana entre muestras fecales completas e hisopos rectales39. En cuanto a la conservación, el almacenamiento inmediato a –80°C se considera el estándar de referencia, aunque ciertos tampones comerciales, como OMNIgene GUT o Zymo DNA/RNA Shield, han demostrado ser adecuados para preservar la estabilidad del microbioma a temperatura ambiente40-42. Otro aspecto importante es el mantenimiento de condiciones anaerobias tras la recogida, ya que la exposición al oxígeno puede alterar significativamente el perfil microbiano43. Además, los protocolos de extracción de ADN influyen notablemente en la representación microbiana, considerando fundamental incluir un paso de disrupción mecánica42,44. Finalmente, la técnica de secuenciación (secuenciación de amplicones o secuenciación shotgun), la plataforma utilizada y el protocolo de preparación de las librerías pueden introducir sesgos relevantes en los resultados obtenidos45.
Puntos críticos en el flujo de trabajo de análisis del microbioma en el que existe variabilidad entre estudios que limitan su comparabilidad e interpretación
| Etapa | Puntos críticos |
|---|---|
| Recogida, conservación y procesado | Tipo de muestra (heces completas, hisopo seco, espátula…) |
| Método y kit de recogida (OMNIgene, Zymo DNA/RNA Shield, etc.) | |
| Tiempo entre recogida y congelación/extracción | |
| Condiciones de conservación y transporte (temperatura, ciclos de congelación-descongelación, anaerobiosis) | |
| Inclusión de metadatos clave (sexo, edad, dieta, medicación, IMC, etc.) | |
| Variabilidad intraindividual y estacional | |
| Extracción de ADN | Carga microbiana fecal (escala de Bristol) |
| Método de lisis (química vs mecánica) | |
| Kit de extracción utilizado (y consistencia entre muestras) | |
| Inclusión de blancos y controles positivos | |
| Rendimiento y pureza del ADN | |
| Centralización del proceso para evitar variaciones entre laboratorios | |
| Secuenciación | Tipo de secuenciación (16S vs metagenómica shotgun) |
| Plataforma de secuenciación empleada (Illumina, Nanopore, etc.) | |
| Preparación de librerías | |
| Profundidad de secuenciación | |
| Coste y disponibilidad | |
| Sesgos por amplificación o cobertura insuficiente | |
| Contaminación cruzada e índice mal asignados («index hopping») | |
| Preprocesamiento de lecturas | Control de calidad (FastQC, Trimmomatic, etc.) |
| Corrección de efectos del lote (ComBat, modelos bayesianos, etc.) | |
| Eliminación de contaminantes (Squeegee, MicrobIEM, etc.) | |
| Ensamblaje vs. mapeo (MEGAHIT, MetaPhlAn, etc.) | |
| Inclusión e interpretación de controles negativos y positivos | |
| Asignación taxonómica y funcional | Herramienta utilizada (QIIME2, DADA2, Kraken2, MetaPhlAn, HUMAnN3, etc.) |
| Base de datos taxonómica (Greengenes, SILVA, ChocoPhlAn, GTDB, etc.) | |
| Base de datos anotación funcional (KEGG, MetaCyc, etc.) | |
| Resolución taxonómica alcanzada (género vs. especie vs. cepa) | |
| Capacidad funcional inferida (predicción vs. metagenómica) | |
| Versiones y actualizaciones de bases de datos | |
| Posibilidad de identificar microorganismos nuevos | |
| Posprocesamiento de datos | Filtro de abundancia y prevalencia aplicados |
| Método de normalización (CPM, CLR, TSS, etc.) | |
| Impacto del «sparsity» (elevada presencia de ceros) | |
| Métodos de reducción de dimensionalidad | |
| Análisis estadístico y clínico | Enfoques aplicados (diversidad, abundancia diferencial, redes, clustering, etc.) |
| Corrección por factores de confusión (dieta, antibióticos, ejercicio, etc.) | |
| Técnica estadística (modelos lineales, mixtos, PERMANOVA, etc.) | |
| Interpretación de resultados en contextos clínicos | |
| Integración con otras ómicas (metabolómica, transcriptómica, etc.) | |
| Aplicación de modelos de inteligencia artificial y machine learning | |
| Limitaciones para inferencias causales |
Por otro lado, también cabe destacar la necesidad de estandarización en las etapas de análisis y tratamiento de los datos. La variabilidad metodológica derivada de las diferencias en el filtrado de secuencias, agrupamiento, asignación taxonómica y binning, debida al uso de distintas herramientas y flujos de trabajo bioinformáticos, introduce sesgos analíticos y estadísticos. Esta heterogeneidad representa una barrera para la reproducibilidad y la comparación entre estudios46. Recientemente, se han comparado múltiples protocolos de análisis metagenómico entre distintos laboratorios, mostrando una considerable variabilidad en los resultados, incluso al procesar las mismas muestras bajo condiciones controladas47.
En este contexto, es especialmente relevante señalar que aún no se dispone de protocolos estandarizados y validados para la evaluación rutinaria de la microbiota intestinal humana en el entorno clínico. Aunque tanto la secuenciación de amplicones del gen 16S del ARNr como la metagenómica shotgun permiten determinar la composición y abundancia de los taxones presentes en una muestra, la falta de estandarización limita su aplicabilidad. La ausencia de herramientas validadas y de biomarcadores clínicamente relevantes impide realizar comparaciones sistemáticas de comunidades microbianas humanas entre diferentes localizaciones o establecer asociaciones robustas con patologías concretas, lo que constituye uno de los principales retos en este campo48.
Concretamente, es relevante destacar que actualmente no se dispone de protocolos estandarizados y validados para la evaluación rutinaria de la microbiota intestinal humana en el contexto clínico. Tanto la secuenciación de amplicones del gen del ARNr 16S como el enfoque shotgun permiten conocer la composición y abundancia de cada taxón en una muestra en la que se quiere caracterizar el microbioma, con la limitación de que el proceso no ha sido estandarizado. Esta falta de herramientas validadas y biomarcadores adecuados impide una comparación más exhaustiva de comunidades microbianas humanas de distinta localización o su asociación con una determinada patología, lo que constituye uno de los retos principales en este campo48.
En este sentido, la elección de la base de datos de referencia también constituye un aspecto crítico que puede influir notablemente en los resultados del análisis del microbioma intestinal. En el caso de la metataxonómica, bases como SILVA13, Greengenes214 o RefSeq15 difieren en su cobertura taxonómica, frecuencia de actualización y criterios de anotación, lo que puede dar lugar a discrepancias sustanciales en los perfiles obtenidos a partir de los mismos datos49. En metagenómica shotgun, la identificación precisa depende aún más de la disponibilidad de genomas completos en bases de datos como RefSeq15 y Genome Taxonomy Database (GTDB)50 o, de forma más específica para el microbioma intestinal humano, el Unified Human Gastrointestinal Genome (UHGG)51 o ChocoPhlAn25. Así, la variabilidad en las bases de datos empleadas y sus constantes actualizaciones constituyen una fuente importante de heterogeneidad entre estudios, subrayando la necesidad de consensuar estándares y emplear recursos especializados según el objetivo y el ecosistema de estudio. Además, una gran proporción de las secuencias obtenidas en estudios metagenómicos sigue sin tener correspondencia con entradas conocidas, lo que limita nuestra capacidad para interpretar el potencial metabólico y clínico del microbioma. En este contexto, el término «materia oscura funcional» se refiere a la gran cantidad de información genómica y funcional que permanece desconocida o no caracterizada en las comunidades microbianas porque no tiene equivalentes conocidos en los organismos previamente estudiados52. Esto incluye secuencias genómicas que no tienen similitudes detectables con aquellas presentes en los genomas de referencia disponibles. Recientemente, Pavlopoulos et al. (2023) han arrojado luz sobre esta materia oscura mediante un enfoque computacional que evita depender de bases de datos de referencia52. Analizando más de 26.000 metagenomas, los autores identificaron más de mil millones de secuencias proteicas sin similitudes conocidas en las bases de datos existentes, lo que resulta en la creación de más de 100.000 nuevas familias de proteínas (novel protein metagenome families [NMPF]), demostrando la magnitud de la diversidad funcional que permanece sin explorar.
Por otro lado, a la hora de implantar el estudio de la microbiota dentro de un laboratorio asistencial, siempre se requiere la estandarización de los procesos, la automatización y sobre todo establecer los puntos de corte para definir la normalidad. Debemos ser conscientes que por el momento no se ha definido cómo es una microbiota «normal» o «saludable» debido a la alta variabilidad individual, tanto en condiciones de salud como de enfermedad. En este contexto, aunque la inclusión de controles positivos y negativos es fundamental, su implementación resulta especialmente compleja por la propia naturaleza del campo y por las dificultades asociadas a su correcta interpretación53. También se necesita una reflexión sobre el uso de las heces como muestra universal para el estudio de la microbiota intestinal. De hecho, las heces contienen los microorganismos que se liberan a la luz intestinal, pero cada vez estamos más seguros de que la organización de los ecosistemas tiene una distribución espacial característica. En cualquier caso, la microbiota del intestino delgado es opuesta a la del intestino grueso, existiendo incluso tramos diferenciales según las condiciones ambientales54. Parece complicado acceder a esos nichos, y como se ha comentado previamente, la funcionalidad es más importante que la composición, pero quizás deberíamos empezar a valorar el paso de metabolitos microbianos a sangre, con potencial efecto sistémico, en lugar de estudiarlos en las heces, o al menos de forma complementaria. A pesar de los esfuerzos por establecer un marco regulador que guíe el uso adecuado del estudio del microbioma y promueva su desarrollo basado en la evidencia, aún existen importantes lagunas de conocimiento y limitaciones que deben resolverse para avanzar en su implementación clínica4.
Métodos estadísticos para el análisis del microbioma y análisis masivo de datosAparte de los retos propios de las técnicas metagenómicas, el análisis de los datos derivados del estudio del microbioma presenta elementos que plantean numerosos desafíos metodológicos. Este tipo de datos se caracteriza por la sobredispersión, lo que significa que las abundancias de las características (i. e. microorganismos, rutas metabólicas, etc.) son altamente variables; una alta dimensionalidad, con potencialmente miles de características perfiladas; y una amplia dispersión, con una elevada presencia de ceros en la matriz de abundancia, a menudo de hasta un 90%. En concreto, esta dispersión ocurre porque muchas especies están presentes en baja abundancia y se encuentran por debajo del umbral de detección del método de secuenciación (ceros técnicos), o porque están completamente ausentes en la muestra (ceros biológicos)55,56. La combinación de estas tres características plantea desafíos para el análisis estadístico y su posterior interpretación.
Centrándonos en los datos metagenómicos, previo al análisis estadístico, los datos crudos se someten a un filtrado adicional con el objetivo de reducir el ruido. En este paso, se filtran las características que presentan una baja abundancia (p. ej.,<500 conteos) y prevalencia (p. ej., <10% de las muestras). También puede elegirse el nivel taxonómico, considerando que descender al nivel de especie conlleva una inflación de ceros significativa57. Además, es importante tener en cuenta la heterogeneidad y la variabilidad entre muestras, por lo que la normalización de los datos de conteo es esencial para mitigar estas variaciones y mejorar la comparabilidad. La normalización es una transformación necesaria para realizar un análisis robusto que considere las peculiaridades de los datos del microbioma y la variabilidad técnica inherente a la tecnología de secuenciación58,59. Entre los métodos más utilizados destacan: Total Sum Scaling (TSS), Cumulative Sum Scaling (CSS), Relative Log Expression (RLE), Aitchison's Log-Ratio (ALR), Aitchison's Centered Log-Ratio (CLR) y Counts Per Million (CPM), y su elección dependerá de la naturaleza de los datos, de la tecnología de secuenciación aplicada, así como del enfoque analítico posterior de los resultados60.
Existen principalmente tres aproximaciones en el estudio del microbioma55, cuyo objetivo es detectar y cuantificar:
- 1.
Taxones diferencialmente abundantes entre grupos de fenotipo (análisis de abundancia diferencial)
- 2.
Asociaciones entre taxones y covariables (análisis integrativo)
- 3.
Asociaciones entre taxones en toda la red del microbioma (análisis de redes)
El método para detectar taxones diferencialmente abundantes entre grupos de fenotipos se conoce como análisis de abundancia diferencial. Esta técnica de análisis permite comprender la relación entre los microorganismos simbióticos y la salud humana, así como identificar biomarcadores microbianos para la detección de enfermedades. Existen numerosos métodos, cada uno con sus propias motivaciones estadísticas. Por ejemplo, edgeR61 surgió de la necesidad de separar la variabilidad biológica de la técnica para reducir el sesgo cuando se buscan diferencias fenotípicas atribuidas a la abundancia de los datos de RNA-Seq. DESeq262 busca un modelo que pueda tener en cuenta la presencia de valores atípicos y tamaños pequeños de réplicas mientras produce resultados interpretables. La dispersión y la inflación con ceros son factores en los que se centran algunos métodos como metagenomeSeq63, Zero-Inflated Beta model (ZIBSeq)64 y Zero-Inflated Generalized Dirichlet-Multinomial model (ZIGDM)65. Analysis Composition of Microbiome (ANCOM)66 surgió por la necesidad de modelos que también pudiesen considerar la naturaleza composicional de los datos de conteo. Además, ZIGDM tiene en cuenta la estructura de correlación y los patrones de dispersión entre las características. Linear Discriminant Analysis Effect Size (LEfSe)67 determina las características que tienen más probabilidad de explicar las diferencias entre grupos al combinar pruebas estándar de significación estadística con pruebas adicionales que codifican la consistencia biológica y la relevancia del efecto de cada característica.
Si bien los métodos mencionados permiten analizar grupos de interés e identificar características asociadas con cada uno, es crucial considerar las múltiples covariables que pueden intervenir, como metabolitos, uso de antibióticos, factores medioambientales y genéticos, etc. La recopilación rigurosa de metadatos, como dieta habitual, uso de medicación, metabolitos, etc., es esencial para interpretar adecuadamente los resultados y controlar potenciales variables confusoras. Por esta razón, surgen los métodos de análisis integrativo, cuyo objetivo es identificar y cuantificar asociaciones entre el microbioma y distintas covariables/metadatos55. Estos métodos proporcionan una visión más completa de las interacciones entre los microorganismos y la salud humana.
El paquete mixOmics68 contiene numerosas herramientas para el análisis multivariante enfocadas en la exploración y reducción de dimensiones de los datos y la visualización de resultados. mixMC69 permite incluir en el análisis tanto datos composicionales como numerosas variables de interés. Además, cuenta con herramientas que permiten integrar 2 o más conjuntos de datos distintos medidos en las mismas muestras, como Data Integration Analysis for Biomarker Discovery using Latent Components (DIABLO)70, y conjuntos de las mismas variables medidas en muestras diferentes, como Multivariate Integrative Method (MINT)71. Estos utilizan técnicas estadísticas de análisis multivariante, como el análisis de componentes principales (Principal Component Analysis [PCA]) y la regresión por proyección de estructuras latentes (Projection to Latent Structures-Discriminant Analysis [PLS-DA]) para seleccionar las características que más discriminan entre grupos.
Las interacciones ecológicas microbianas afectan la función del microbioma y la salud del hospedador a través de la formación de comunidades complejas con relaciones simbióticas donde los microorganismos coexisten. El objetivo del análisis de redes es construir redes de microbiomas que caractericen las asociaciones ecológicas microbianas, lo que puede ayudar a descubrir propiedades y mecanismos fundamentales de los ecosistemas microbianos55. Los modelos gráficos consisten en nodos y aristas que se utilizan para visualizar la red microbiana estimada. Cada nodo corresponde a un taxón y una arista existente representa una asociación directa entre 2 nodos cualesquiera. Los métodos estadísticos actuales para el análisis de redes estiman la estructura de correlación, como Sparse Correlations for Compositional data (SparCC)72 o correlación parcial, como Hybrid Approach foR MicrobiOme Network Inferences via Exploiting Sparsity (HARMONIES)73, de los datos de conteo normalizados para construir una red de nodos y aristas (tabla 3).
Métodos estadísticos para el análisis del microbioma
| El análisis de datos del microbioma plantea retos únicos debido a su alta dimensionalidad, dispersión y sobredispersión. Estos desafíos requieren transformaciones iniciales y enfoques estadísticos específicos: |
| 1. Filtrado y normalización de datos: Los datos crudos suelen someterse a un filtrado adicional por baja abundancia y/o prevalencia para disminuir el ruido. Además, métodos de normalización como TSS o CLR se emplean para mitigar la variabilidad técnica y biológica |
| 2. Análisis de abundancia diferencial: Métodos como DESeq2, LEfSe y ANCOM permiten identificar taxones y funciones diferencialmente abundantes entre grupos fenotípicos |
| 3. Análisis integrativo: herramientas como MINT o DIABLO integran datos del microbioma con covariables ambientales, genéticas y metabólicas |
| 4. Análisis de redes: métodos como SparCC o HARMONIES identifican interacciones ecológicas entre microorganismos, revelando propiedades clave |
Los métodos estadísticos clásicos se han ido remplazando por otros más sofisticados, como herramientas de inteligencia artificial (IA). La IA y, en particular, el aprendizaje automático (machine learning [ML]) y el profundo (deep learning) están abriendo nuevas fronteras en la investigación del microbioma (tabla 4). El uso de estas herramientas puede mejorar la comprensión, el diagnóstico y el tratamiento de enfermedades asociadas al microbioma humano74. La IA permite analizar conjuntos de datos complejos y de gran volumen mediante la identificación de patrones y asociaciones limitados en otros métodos estadísticos tradicionales. Asimismo, facilita el análisis de datos metagenómicos, mejorando la identificación y la clasificación de especies microbianas. Estas herramientas pueden utilizarse para diseñar modelos predictivos y mejorar la precisión diagnóstica de algunas enfermedades asociadas al microbioma. Además, pueden contribuir a los tratamientos personalizados al mejorar el conocimiento de los perfiles individuales. Si bien requiere una mayor cantidad de datos robustos y comparables de lo que los estudios de microbiota suelen disponer, la implementación de estándares en la recolección y análisis de datos metagenómicos, como los propuestos por proyectos como el American Gut Project (https://www.ebi.ac.uk/metagenomics/studies/MGYS00000596#overview), facilitan la generación de datos de alta calidad para el entrenamiento de modelos predictivos75.
Inteligencia artificial (IA) y aprendizaje automático
| El uso de IA y aprendizaje automático ha revolucionado el análisis del microbioma, facilitando la detección de patrones complejos y el desarrollo de modelos predictivos. Aplicaciones comunes incluyen: |
| ML no supervisado: técnicas como la agrupación jerárquica y PCA para identificar estructuras de datos y patrones |
| ML supervisado: métodos como RF y SVM para clasificar y predecir características del hospedador |
| Estas herramientas están transformando el campo hacia la identificación de biomarcadores, la personalización de tratamientos y el desarrollo de intervenciones preventivas. Sin embargo, persisten retos relacionados con la calidad de los datos, la estandarización de protocolos y el manejo ético de la IA |
El ML es un subconjunto de métodos de IA utilizado para reconocer, clasificar y predecir patrones. En la investigación del microbioma, el ML se ha aplicado a tareas como la predicción del fenotipo del hospedador, la clasificación de características microbianas (es decir, determinar la abundancia, la diversidad o la distribución de la microbiota), el estudio de las complejas interacciones fisicoquímicas entre los componentes del microbioma, y el seguimiento de los cambios en su composición74,76. Los modelos ML pueden entrenarse para predecir la composición de las comunidades microbianas en función de varios factores de entrada, como la genética del hospedador, la dieta y los factores ambientales, lo que puede ayudarnos a comprender los factores que influyen en la composición microbiana y su relación con la salud humana57.
El análisis de datos mediante ML puede abordarse desde dos perspectivas principales. Los métodos de ML no supervisados tratan de buscar patrones en conjuntos de datos sin variables dependientes o etiquetas conocidas. Estas técnicas permiten realizar dos aproximaciones diferentes: el agrupamiento o clustering y la reducción de la dimensionalidad77. Las técnicas de clustering organizan las muestras en grupos basándose en medidas de similitud. Dentro de estas, destacan la agrupación jerárquica o hierarchical clustering, que construye una jerarquía mediante la combinación o división de los grupos según una medida de disimilitud y la agrupación k-means, que divide los datos en un número predeterminado de grupos, denotados como k, asignando cada observación a un grupo de acuerdo con su distancia al centro de ese grupo78. Estas herramientas han permitido la identificación de patrones novedosos en el estudio del microbioma intestinal, como el descubrimiento de enterotipos o grupos de genes de coabundancia79. Por otro lado, las técnicas de reducción de dimensionalidad permiten representar datos con una elevada dimensionalidad (es decir, con un gran número de variables) a partir de la extracción de las variables más relevantes. Entre ellas, se incluyen el PCA, basado en una matriz de covarianza y la distancia euclidiana, y el análisis de coordenadas principales (Principal Coordinate Analysis), que utiliza otras métricas de disimilitud, como aquellas aplicadas en el análisis de la β-diversidad77.
Los métodos de ML no supervisados son herramientas exploratorias útiles para examinar los datos y determinar estructuras de datos importantes y patrones de correlación78. Sin embargo, los métodos de ML supervisados son más comúnmente utilizados en la predicción de características del hospedador. Para ello, primero se entrena un modelo a partir de datos o características de entrada (variables independientes) complementados con variables dependientes o etiquetas que indican los resultados para las muestras de entrada. El modelo generado es potencialmente capaz de predecir los resultados de muestras nuevas. Cuando las variables dependientes son categóricas, el modelo de ML se puede emplear en tareas de clasificación, mientras que, si son numéricas continuas, pueden realizar tareas de regresión79. Entre los modelos de ML supervisado más utilizados en el análisis del microbioma destacan los modelos de regresión (lineal o logística), el análisis discriminante lineal (Linear Discriminant Analysis [LDA]), el bosque aleatorio (Random Forest [RF]), las máquinas de vectores de soporte (Support Vector Machines) y las redes neuronales80. Particularmente, los métodos de aprendizaje en conjuntos basados en árboles de decisión, como el RF, han sido ampliamente aplicados en estudios sobre el microbioma intestinal79,81.
Hasta la fecha, existen numerosas herramientas estadísticas para el estudio del microbioma. No obstante, las transformaciones iniciales y la elección del método estadístico dependen en gran medida de los datos disponibles y de los objetivos específicos del estudio. Además, el campo del microbioma está evolucionando desde la identificación de asociaciones hacia la búsqueda de causalidad y predicción, donde las herramientas de ML jugarán un papel crucial en esta transición. Por otro lado, para promover la investigación y el trabajo sobre la identificación de características «ómicas» predictivas y discriminatorias, es necesario mejorar la repetibilidad y la comparabilidad, desarrollar procedimientos de automatización y definir áreas prioritarias para el desarrollo de nuevos métodos de aprendizaje automático dirigidos al microbioma82. Sin embargo, y aunque el potencial de la aplicación de la IA en la investigación de la microbiota es extraordinario, también existen importantes retos relacionados con la calidad y la normalización, la mejora de los algoritmos que manejen mejor la variabilidad y complejidad de los datos del microbioma, su combinación con otros datos genómicos, proteómicos y metabolómicos, y con garantizar su uso ético en investigación83.
Conclusiones y perspectivas futuras en el estudio del microbioma intestinalEl estudio del microbioma intestinal ha avanzado significativamente, gracias a las tecnologías de secuenciación de nueva generación y a las herramientas bioinformáticas. Sin embargo, todavía enfrenta desafíos críticos que limitan su máximo potencial, especialmente en aplicaciones clínicas y en ámbitos como la nutrición y la salud. La ausencia de protocolos estandarizados para la recolección, almacenamiento, extracción y análisis de muestras introduce una alta variabilidad técnica que dificulta la reproducibilidad de los estudios y la comparación entre laboratorios. La implementación de guías internacionales que armonicen estos procedimientos es imperativa para superar estas barreras y construir una base sólida para el desarrollo de aplicaciones biomédicas, particularmente en el diseño de intervenciones personalizadas y estrategias de prevención basadas en la modulación del microbioma.
A pesar de estas limitaciones, los avances en la integración de datos multiómicos y el uso de tecnologías como la IA y el aprendizaje automático, están transformando la forma en que abordamos el microbioma. Estas herramientas no solo permiten identificar patrones complejos, sino que facilitan la predicción de interacciones microbioma-hospedador y la identificación de biomarcadores clave. En el ámbito clínico, estas innovaciones abren nuevas posibilidades, como la personalización basada en el perfil microbiano individual, la identificación de componentes dietéticos, fármacos y terapias que modulan el microbioma, y el diseño de estrategias terapéuticas dirigidas.
La normalización de los procedimientos para el análisis del microbioma es fundamental para que investigadores y profesionales puedan interpretar con confianza los resultados a lo largo del tiempo, así como para facilitar las comparaciones entre individuos de un mismo estudio o cohorte. Este artículo presenta los principales puntos críticos en el flujo de trabajo relativo al análisis y la gestión de datos relacionados con el microbioma intestinal, donde es necesaria la estandarización (fig. 2). La complejidad de los datos microbiológicos también plantea retos importantes, como la necesidad de mejorar la interpretación funcional de la materia oscura genómica y garantizar la calidad de los datos utilizados en algoritmos de IA. Abordar estas limitaciones será esencial para que el microbioma intestinal se convierta en una herramienta central en la prevención y tratamiento de enfermedades. Además, consolidará su papel en la próxima generación de terapias, estrategias de salud pública y enfoques innovadores que promuevan un envejecimiento saludable y la reducción de enfermedades crónicas relacionadas con el estilo de vida.

Paralelamente, el manejo ético de los datos generados en el estudio del microbioma adquiere una relevancia creciente. Es necesario garantizar la privacidad y seguridad de los datos y garantizar un acceso equitativo a sus aplicaciones en salud. La creación de marcos regulatorios que integren estas consideraciones será clave para fomentar la confianza pública y maximizar el impacto positivo del microbioma en la salud global.
Finalmente, la colaboración interdisciplinaria y el intercambio de datos entre investigadores, instituciones y países serán fundamentales para acelerar el desarrollo de nuevas aplicaciones clínicas y nutricionales. Solo mediante un enfoque global y coordinado será posible superar los desafíos actuales y aprovechar plenamente las oportunidades que ofrece el estudio del microbioma para mejorar la salud humana.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores agradecen la financiación recibida del Ministerio de Ciencia, Innovación y Universidades (Proyecto PID2023-148419OB-I00, MICIN). Agradecemos el apoyo de la Red Científica Conexión-MICROBIOMA, financiada por el Consejo Superior de Investigaciones Científicas (CSIC), así como la Red-MIDAS (RED2022-134934-T) financiada por el MICIN.

