








Chlamydophila pneumoniae es un patógeno humano intracelular, muy prevalente, con un ciclo único de desarrollo bifásico, que causa infecciones respiratorias en las vías altas y neumonía, y que actualmente se cree que puede ser un factor de riesgo para el desarrollo de la arteriosclerosis.
C. pneumoniae muestra una gran complejidad en los antígenos de superficie de la membrana externa, ya sea por ser específicos pero poco inmunógenos (como la proteína principal de la membrana externa) o bien por ser muy inmunógenos pero poco específicos entre las especies de clamidias. Todo esto hace necesario profundizar en el estudio de nuevos antígenos que sean altamente inmunodominantes y específicos de especie. En este sentido, las proteínas polimórficas de la membrana externa (PMP) son a) específicas de clamidia, b) se exponen en la superficie de la bacteria y c) son muy inmunógenas, todo lo cual las hace acreedoras de un importante potencial de aplicación en los diferentes ensayos de laboratorio. Otras, como la proteína de choque térmico 60 (HSP 60), parecen estar relacionadas con la arteriosclerosis, por inducir un ataque inmunológico sobre la pared endotelial.
A partir de los estudios existentes hasta la fecha, para obtener una conclusión sobre la relación entre la infección y la arteriosclerosis, faltan estudios con suficiente número de pacientes y muestras, prospectivo, comparativo frente a controles sanos, que utilicen la combinación de varias técnicas microbiológicas (directas e indirectas) en un mismo sujeto y muestra, relacionando los resultados con la actividad de la enfermedad.
Chlamydophila pneumoniae is a highly prevalent intracellular human pathogen with a unique biphasic life cycle. It is a common cause of upper respiratory infection and pneumonia, and is currently being studied as a potential risk factor for the development of atherosclerotic cardiovascular disease.
The outer membrane surface antigens of C. pneumoniae are highly complex: some, such as the major outer membrane protein, are specific, but poorly immunodominant, whereas others have stronger immunogenicity, but are cross-reactive among Chlamydia species. Therefore, new, highly immunodominant, species-specific antigens should be sought. In this regard, the polymorphic membrane proteins (PMPs) are a) unique to Chlamydiae, b) often exposed on the surface of the bacteria, and c) highly immunogenic; these factors make them potential candidates for application in laboratory assays. Other chlamydial antigens, such as heat shock protein (HSP) 60, have been associated with atherosclerotic lesions because of their ability to induce an immunological attack on the endothelial wall.
Over the last decade, several studies have suggested a potential role of chronic C. pneumoniae infection in human atherosclerosis. Nevertheless, prospective studies with sufficiently large samples and a healthy comparison group, using a combination of direct and indirect microbiological techniques in the same subject and sample, are needed to establish a relationship between the infection and disease activity.
Históricamente, el orden Chlamydiales incluye la familia Chlamydiaceae, con un solo género, Chlamydia, y cuatro especies: Chlamydia trachomatis, Chlamydia psittaci, Chlamydia pneumoniae y Chlamydia pecorum. Más recientemente, a partir del análisis de las secuencias de los genes del ARNr 16S y 23S, se comprobó que existían diferencias entre C. trachomatis y el grupo C. psittaci-C. pneumoniae, lo que llevó a dividir esta familia en dos géneros diferentes1: Chlamydia y Chlamydophila, respectivamente. No obstante, junto con los géneros anteriores, patógenos humanos, existen otros, pertenecientes al orden Chlamydiales, que carecen, en estos momentos, de interés clínico.
Chlamydophila pneumoniae es muy conocido actualmente por ser un agente productor de enfermedad respiratoria aguda, incluyendo neumonía, bronquitis, sinusitis y faringitis y, así como por las investigaciones que la relacionan con la arteriosclerosis, entre otras enfermedades. Este organismo fue aislado por primera vez en 1965, de la conjuntiva de un niño taiwanés durante la vacunación antitracoma2.
Ciclo celularDurante su ciclo celular adopta dos morfologías distintas, una forma infecciosa extracelular, el cuerpo elemental, y una forma replicativa intracelular, el cuerpo reticular. El primero es pequeño y denso, tiene un tamaño de entre 0,2 y 0,4 μm, y una forma característica de pera. Morfológicamente, es distinto de los cuerpos elementales redondeados de C. trachomatis y C. psittaci. No obtiene nutrientes del exterior y carece de actividad metabólica y de replicación. Posee una pared celular rígida, aunque lo suficientemente laxa como para permitir esta forma de pera, gracias a proteínas ricas en aminoácidos azufrados, que forman un entramado denso, que le hace resistente a los factores ambientales.
El cuerpo reticular es más grande, con un tamaño de entre 0,6 y 1,2 μm. Procede de la transformación del cuerpo elemental tras entrar en la célula hospedadora. Éste obtiene nutrientes del citoplasma de la célula y se multiplica por división binaria. Los cuerpos reticulares son lábiles, osmóticamente inestables e incapaces de infectar a otras células.
El ciclo celular se inicia cuando un cuerpo elemental, por su zona más aguda, se une a una célula por un mecanismo del tipo adhesina-receptor (fig. 1A). Si bien la identidad precisa de ambas moléculas continúa siendo incierta, parece tratarse de un sistema del tipo glucosamino-glucano parecido al sulfato de heparina. Penetran en la célula por endocitosis y se forma un fagosoma, pero no un fagolisosoma3. Es característica del orden de las Chlamydiales su habilidad para inhibir la fusión lisosomal, por mecanismos no definidos, permitiendo al cuerpo elemental habitar en una vesícula, rodeada de una membrana protectora, llamada cuerpo de inclusión, visible al microscopio óptico (fig. 1B). Entonces, se modifica la membrana externa del cuerpo elemental, desaparecen los aminoácidos azufrados y se conforman, funcionalmente, las porinas.

El paso de cuerpo elemental a cuerpo reticular, y viceversa, requiere un ciclo intracelular de 2-4 días en C. pneumoniae. El cuerpo elemental se transforma en reticular cuando penetran metabolitos, como fosfatos ricos en energía, y aminoácidos, aumentando la actividad metabólica. Estos procesos iniciales incluyen la síntesis de nuevas proteínas, reducción de los enlaces disulfuro y la activación de la adenosina trifosfatasa4.
Los cuerpos reticulares se dividen de forma binaria e incesante, agrupados en el fagosoma (fig. 1C). El cuerpo de inclusión y la célula infectada muestran en la superficie antígenos derivados de la bacteria. Los cuerpos reticulares madurarán, reduciendo su tamaño y reorganizando su pared celular, a modo de cuerpos elementales. En ese momento, los cuerpos de inclusión contendrán cuerpos reticulares maduros, futuros elementales (fig. 1D).
La liberación de los cuerpos elementales de las células infectadas se puede realizar por lisis celular, extrusión o exocitosis, y se cierra el ciclo de desarrollo permitiendo la infección de nuevas células (fig. 1E).
Por determinadas condiciones ambientales, o dependiendo del estado del agente infeccioso o de la célula hospedadora, el ciclo se puede detener, permanente o temporalmente, en la fase de cuerpo reticular, que se denomina ahora cuerpo persistente (fig. 1C'). Así, Beatty et al5, realizando cultivos celulares en líneas de epitelio de faringe humana, determinaron que uno de los mecanismos que induce a entrar a la bacteria en un estado persistente es la presencia de interferón gamma (IFN-γ).
Estos cuerpos persistentes son cuerpos reticulares más grandes, que, por el menor número de porinas, se dividen lentamente. Este estado críptico conlleva la reducción en la expresión de antígenos, cambios de la morfología y la pérdida de la infectividad. Se piensa que estas formas podrían tener un papel importante en la patogénesis de la infección crónica, debido, entre otros, a la presencia en este estadio de proteínas de choque térmico (heat shock protein [HSP])6. Esta fase puede ser temporal y luego continuarse el ciclo, como antes se ha comentado.
Los cuerpos persistentes podrían ser una adaptación de la bacteria al hospedador para evitar el sistema inmunológico, reducir una virulencia innecesaria o mantenerse viable en condiciones adversas. Su presencia podría tener, como en el caso de C. trachomatis7, repercusiones diagnósticas (ya que determinarían falsos negativos en el inmunodiagnóstico, por la escasa expresión de antígenos bacterianos, o en el cultivo celular, por la menor viabilidad del cuerpo persistente), y terapéuticas (por una menor respuesta al tratamiento debido a la pérdida de porinas y la menor expresión de dianas).
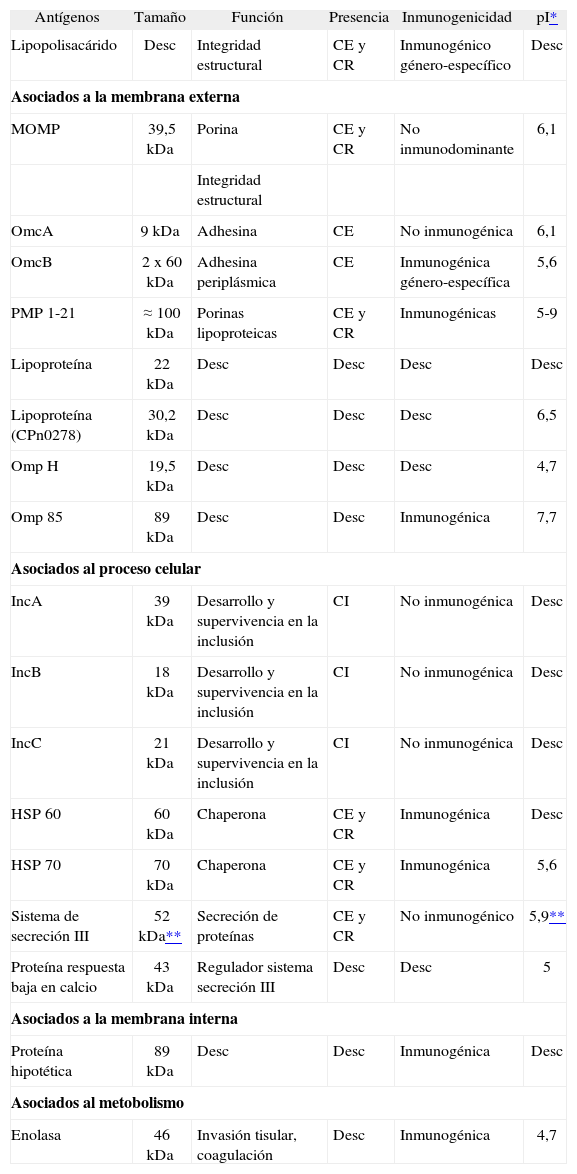
Antígenos de C. pneumoniaeLos antígenos de C. pneumoniae son complejos y sólo parcialmente conocidos. Las primeras descripciones fueron realizadas por Kuo et al2. Se localizan en la pared celular (fig. 2), y, entre otros, destacan los siguientes grupos (tabla l)8:

Características de los antígenos descritos en C. pneumoniae
| Antígenos | Tamaño | Función | Presencia | Inmunogenicidad | pI* |
| Lipopolisacárido | Desc | Integridad estructural | CE y CR | Inmunogénico género-específico | Desc |
| Asociados a la membrana externa | |||||
| MOMP | 39,5 kDa | Porina | CE y CR | No inmunodominante | 6,1 |
| Integridad estructural | |||||
| OmcA | 9 kDa | Adhesina | CE | No inmunogénica | 6,1 |
| OmcB | 2 x 60 kDa | Adhesina periplásmica | CE | Inmunogénica género-específica | 5,6 |
| PMP 1-21 | ≈ 100 kDa | Porinas lipoproteicas | CE y CR | Inmunogénicas | 5-9 |
| Lipoproteína | 22 kDa | Desc | Desc | Desc | Desc |
| Lipoproteína (CPn0278) | 30,2 kDa | Desc | Desc | Desc | 6,5 |
| Omp H | 19,5 kDa | Desc | Desc | Desc | 4,7 |
| Omp 85 | 89 kDa | Desc | Desc | Inmunogénica | 7,7 |
| Asociados al proceso celular | |||||
| IncA | 39 kDa | Desarrollo y supervivencia en la inclusión | CI | No inmunogénica | Desc |
| IncB | 18 kDa | Desarrollo y supervivencia en la inclusión | CI | No inmunogénica | Desc |
| IncC | 21 kDa | Desarrollo y supervivencia en la inclusión | CI | No inmunogénica | Desc |
| HSP 60 | 60 kDa | Chaperona | CE y CR | Inmunogénica | Desc |
| HSP 70 | 70 kDa | Chaperona | CE y CR | Inmunogénica | 5,6 |
| Sistema de secreción III | 52 kDa** | Secreción de proteínas | CE y CR | No inmunogénico | 5,9** |
| Proteína respuesta baja en calcio | 43 kDa | Regulador sistema secreción III | Desc | Desc | 5 |
| Asociados a la membrana interna | |||||
| Proteína hipotética | 89 kDa | Desc | Desc | Inmunogénica | Desc |
| Asociados al metobolismo | |||||
| Enolasa | 46 kDa | Invasión tisular, coagulación | Desc | Inmunogénica | 4,7 |
CE: cuerpo elemental; CI: cuerpo de inclusión; CR: cuerpo reticular; Desc: desconocido. HSP: proteína de choque térmico; Inc: proteínas de inclusión; PMP: proteínas polimórficas de membrana externa.
El lipopolisacárido es un antígeno presente en todos los miembros de la familia Chlamydiaceae. Su presencia se ha demostrado mediante fijación del complemento, inmunofluorescencia y ELISA9,10 y es una causa importante de las reacciones cruzadas que, con frecuencia, se observan en la serología de Chlamydiaceae.
Está presente en los cuerpos elementales, en los reticulares, en la membrana de las células infectadas y en la región proximal de células anejas a la anterior, pero no infectadas. Su presencia se ha demostrado a las 20 h de la infección celular. Dado que la superficie de los cuerpos reticulares es 10 veces superior a la de los elementales, durante la transformación de aquéllos, es probable que sean expulsadas al exterior grandes cantidades de lipopolisacárido a través de vesículas, desde la inclusión intracelular, al citoplasma y a la superficie de la célula. Este hecho apoya la hipótesis que implica al lipopolisacárido en la patogenia de las manifestaciones clínicas asociadas a C. pneumoniae11.
De manera indirecta, a partir de la capacidad de neutralización de los anticuerpos monoclonales, pero no bien caracterizada, se han supuesto también, en el lipopolisacárido de C. pneumoniae, la presencia de epítopos específicos de especie12. Del mismo modo, los estudios con anticuerpos han sugerido que, durante el proceso de transformación de los cuerpos reticulares en elementales, la estructura del lipopolisacárido puede sufrir algún cambio9.
Proteínas de la membrana externaProteína principal de la membrana externa (MOMP)Uno de los antígenos mejor caracterizados en las especies de Chlamydia es la MOMP. Es la proteína que se halla en mayor proporción (60%) en los cuerpos elementales y reticulares10.
Tiene funciones de porina. Para ello, se debe encontrar químicamente reducida, situación que modifica las uniones entre las proteínas ricas en cisterna y que permite dicha función13 en los cuerpos reticulares10 activos metabólicamente.
Esta proteína es común a todos los miembros de la familia Chlamydiaceae, aunque presenta diferencias estructurales y de inmunogenicidad en cada una de las especies. Tiene cuatro dominios variables, que tienden a situarse en la superficie de la membrana, y cinco constantes. La secuencia, en C. pneumoniae, no se considera inmunodominante. Esto se ha intentado explicar por la supuesta incapacidad del sistema inmunológico para identificar los epítopos superficiales de la MOMP, a causa de un mecanismo de interposición o enmascaramiento que ejercerían las proteínas polimórficas de la membrana externa (PMP)14. Esto no impide que algunos anticuerpos monoclonales sí puedan reconocer antígenos especie-específicos de C. pneumoniae y neutralizar la infección in vitro15.
Proteínas del complejo de membrana externa (OMC) ricas en cisteínaEstán presentes en grandes cantidades en el cuerpo elemental y se sintetizan de manera tardía en la maduración del cuerpo reticular hacia el elemental. No están presentes en el cuerpo reticular (a diferencia de la MOMP)16.
La proteína OmcA recuerda a las lipoproteínas mureínicas de las bacterias gramnegativas. Es una adhesina que se fija a las moléculas de heparina, facilitando la infectividad. Está anclada en la membrana externa por su región lipídica, mientras que la zona peptídica se extiende hacia el periplasma17.
Más controvertida es la estructura de la OmcB18. Se sabe que la posición de las cisteínas es muy mantenida y es muy inmunogénica, pero comparte epítopos con otras especies bacterianas. Finalmente, los puentes disulfuro de OmcB forman uniones cruzadas con los dominios periplásmicos de OmcA y de otras proteínas, contribuyendo a la estabilidad del cuerpo elemental17. Algunos autores proponen que este complejo es el equivalente del peptidoglucano, cuya presencia en Chlamydia es discutida19.
Proteínas polimórficas de la membrana externa (PMP)Existen hasta 21 tipos en C. pneumoniae. Su superficialidad e inmunogenicidad justifican el interés por estas proteínas20. Son ricas en serina y fenilalanina, y son polimórficas, ya que sus genes sufren mutaciones que originan variaciones en la estructura21. Se piensa que dicha variabilidad depende del número de residuos de guanina, y no está claro si se debe a la presión del sistema inmunológico, o es intrínseca. Biológicamente, actúan como citolisinas, que contribuyen a la rotura de la célula hospedadora y a la salida del cuerpo elemental, y, además, posiblemente, tienen un papel en la entrada al hospedador22.
Otras proteínas de la membrana externaOtras proteínas presentes en C. pneumoniae, y cuya función no es bien conocida, se reflejan en la tabla 1.
Proteínas del proceso celularProteínas de inclusión (Inc)Se localizan en la membrana de los cuerpos de inclusión de C. pneumoniae y el resto de miembros del orden Chlamydiales. Cada proteína posee un dominio hidrofóbico único de 50 a 80 aminoácidos23. Sus funciones se han relacionado con el desarrollo de la inclusión, la evasión del sistema inmunológico, la adquisición de nutrientes e, incluso, son mediadores en los procesos de transición de cuerpos reticulares a elementales, y viceversa. Constituyen una familia de proteínas que tienen un importante papel en la infección, el crecimiento y la supervivencia en el hospedador celular24.
ChaperonasParte de ellas son HSP y corrigen plegamientos incorrectos de proteínas desnaturalizadas. La HSP 60 parece estar implicada en los daños ocasionados en las arterias de los sujetos infectados debido a la hiperestimulación de macrófagos y a la inducción de los autoanticuerpos, por mimetismo molecular con la proteína humana25, citocinas proinflamatorias y metaloproteinasas, que degradarían el colágeno26. Se relaciona, por tanto, con la arteriosclerosis, por inducir un ataque a la pared endotelial, mediante una reacción de hipersensibilidad retardada, propia de las infecciones crónicas27, que puede ser el eslabón entre los microorganismos involucrados y la autoinmunidad28.
Por su parte, la HSP 70 está presente de forma temprana en la infección y tiene un importante papel en el transporte de proteínas a través de las membranas29.
Sistema de secreción tipo III y proteínas de respuesta baja en calcioLas características30,31 de estas proteínas presentes en C. pneumoniae, y cuya función no es bien conocida, se reflejan en la tabla 1.
Proteínas relacionadas con el metabolismo32Entre las proteínas relacionadas con el metabolismo energético está la enolasa. Esta proteína de la superficie bacteriana comparte antígenos con la enolasa humana, presente en las células hematopoyéticas, por lo que, en las infecciones por clamidia, la inducción de anticuerpos antienolasa podría causar reacciones autoinmunes inflamatorias8,33.
PeptidoglucanoSu presencia se ha sospechado por estudios, inicialmente referidos a los cuerpos reticulares de C. trachomatis, en los que se comprobó que antibióticos como penicilina o cicloserina inhibían el proceso de división celular34. Asimismo, se ha demostrado la presencia, en el genoma del serovar D de C. trachomatis, de genes para la síntesis del peptidoglucano35. Sin embargo, los estudios realizados con objeto de detectar ácido N-acetilmurámico, la molécula característica del peptidoglucano, no han sido concluyentes. Entonces, es posible que el peptidoglucano esté presente, de forma transitoria y escasa, en determinados momentos del ciclo vital, y con funciones muy concretas. Apoyando esta hipótesis se ha sugerido que, en el cuerpo reticular, sustituiría la función de las proteínas ricas en cisteína de la pared celular del CE, participando en la división de la bacteria36, y en la adhesión a la membrana y superficie celular37.
Patogenia de la infección por C. pneumoniaePara penetrar en el organismo humano la bacteria utiliza, entre otras, la célula epitelial columnar o de transición, infectando las vías respiratorias y los monocitos. Inicialmente lo hace en las células respiratorias de las vías altas y/o bajas y se disemina por la sangre gracias a los monocitos. Esto le permite la colonización a distancia de muchos lugares del organismo, tales como las arterias. Tras la infección inicial del epitelio respiratorio de las vías altas el sujeto puede quedar como portador debido a un fracaso en la actuación de la respuesta inmunológica38.
Al igual que con C. trachomatis, las personas infectadas por C. pneumoniae desarrollan una respuesta inmunológica celular39. Ésta es la más importante, suele ser poco intensa, pero persistente en el tiempo, y puede resolver la infección.
La bacteria, durante la infección de la célula hospedadora, detiene la apoptosis, lo que contribuye a la replicación y facilita una posible cronificación. Las células T CD4 son activadas por los antígenos de Chlamydia, que derivan del contenido de las vacuolas, y se relacionan con las moléculas de clase II del sistema HLA, que se exponen en las células infectadas como macrófagos, dendritas y células endoteliales; el reconocimiento de los antígenos producirá la secreción de citocinas. Éstas activan los macrófagos y las células B para producir anticuerpos6.
El IFN-γ inhibe la replicación de los patógenos intracelulares estrictos, por inducción de la producción de indolamina 2,3-dioxigenasa (IDO). La IDO degrada el triptófano a quineurina y N-formilquineurina, deprivando al patógeno de triptófano. Como resultado, el crecimiento bacteriano puede ser restringido. Cuando el triptófano es repuesto, la inhibición se revierte40. Carlin y Weller41 investigaron algunos agentes inmunomoduladores que podían actuar sinérgicamente con los IFN, en la restricción del crecimiento de Chlamydia por incremento de la actividad de la IDO. La adición de interleucina 1 (IL-1) aumentó la producción de IDO y suprimió el crecimiento de Chlamydia. La IL-1β parece ser la más importante. Entonces es posible que un déficit de IFN-γ sea el causante de la aparición de una infección persistente, a través de la menor inducción de la IDO (v. ciclo replicativo en fig. 1).
La reinfección es frecuente a lo largo de la vida porque surgen variantes alélicas, debido a la presión inmunológica o recombinación genética en la infección mixta. Además, no se ha demostrado la capacidad de neutralización de los anticuerpos para evitar nuevas infecciones15. Finalmente, hay evidencias de que una infección reciente superada puede conferir protección frente a enfermedades graves posteriores, aunque no a la reinfección42.
El período de incubación de la enfermedad aguda es de varias semanas (media de tres), más largo que para otros muchos patógenos respiratorios43. A través de la investigación de los anticuerpos, generalmente mediante el empleo de pruebas de microinmunofluorescencia44, se ha demostrado que la infección tiene una presencia universal45, que los sujetos se infectan alguna vez en su vida y que la seroprevalencia es más importante a medida que aumenta la edad debido a las reinfecciones repetidas.
La bacteria es capaz de originar infecciones asintomáticas en niños, por infección primaria. Esto no excluye la posibilidad de ocasionar faringitis, otitis y sinusitis. Así, la bacteria se ha asociado con sinusitis purulenta46, otitis media supurada47 y faringitis, pero se desconoce la frecuencia real de estas enfermedades48.
Las infecciones sintomáticas suelen ocurrir en adultos, debido a reinfección, y menos frecuentemente por reactivación. Así, ocasiona bronquitis y neumonía (hasta el 20% de las neumonías extrahospitalarias ocurridas en los mayores de 65 años y el 5% de los casos de bronquitis y sinusitis) y es una causa de reactivación clínica de los sujetos con enfermedad pulmonar obstructiva crónica (EPOC)38.
Más recientemente, se ha asociado con otros procesos de etiología hasta ahora no bien conocidos, tales como la arteriosclerosis.
C. pneumoniae y su relación con la arteriosclerosisActualmente, el área de investigación más activa en relación con este patógeno es la posible influencia de la infección vascular por C. pneumoniae en la patogénesis de las enfermedades cardiovasculares arterioscleróticas. Esto se debe a que los factores de riesgo clásicos para la arteriosclerosis sólo explican el 60% de los casos, y nuevos agentes, tales como C. pneumoniae, se han asociado con la enfermedad49.
Desde hace años se han obtenido muchas evidencias microbiológicas de la presencia de C. pneumoniae en las placas de ateroma de las arterias coronarias, carótida, aorta, ilíaca y femoral, mediante visualización con microscopía electrónica o inmunohistoquímica y pruebas de PCR50–52.
Observaciones procedentes de la infección experimental de conejos sugieren que la infección por C. pneumoniae puede facilitar el desarrollo de placas de ateroma en animales hipercolesterolémicos. La lesión inicial inducida por el colesterol podría ser necesaria para que pueda infectar y residir en la arteria53 o una consecuencia de la infección. Esto se apoya en el hecho de que la infección induce, in vitro, un marcado aumento de la captación de ésteres de colesterol por los monocitos humanos incubados en presencia de lipoproteínas de baja densidad (LDL), de modo que éstos acaban evolucionando a células espumosas54. Entonces ¿cómo se podría producir la placa de ateroma en el curso de la infección?
Resumiendo los conocimientos existentes hasta la fecha, podríamos establecer una secuencia de los acontecimientos (figs. 3 y 4). Se sabe que los vasos se pueden lesionar por: una concentración elevada de LDL y/o una alteración de ésta; la presencia de radicales libres causados por el tabaco, hipertensión o diabetes mellitus; alteraciones genéticas; agentes infecciosos como herpesvirus o C. pneumoniae; concentraciones plasmáticas elevadas de homocisteína, o una combinación de cualquiera de estos factores55. Por lo expuesto anteriormente se sabe que C. pneumoniae puede infectar, en el vaso, el endotelio, previamente lesionado por cualquiera de las causas anteriores, el músculo liso y los macrófagos, con más facilidad en presencia de grasas. La infección de las células endoteliales permitiría liberar cuerpos elementales al medio extracelular, que servirían para extender la infección localmente, o por vía sistémica4,56.
. (1) Lesión de los vasos por diferentes causas. (2) Esta lesión incrementa la síntesis de proteína quimiotáctica 1 de monocito y moléculas de adhesión que fijan leucocitos infectados; actividad procoagulante de lipoproteínas tisulares, con trombosis y adhesión de plaquetas. La célula endotelial se infecta por C. pneumoniae intramonocítica. (3) Los monocitos migran al subendotelio, liberan citocinas proinflamatorias y expresan la molécula CD14. El interferón 7 aumenta la dioxigenasa intramonocítica que oxida el triptófano hasta quineurina. (4) Se infectan las células musculares lisas y (5) los macrófagos.")
Patogenia de la arteriosclerosis y su relación con la infección por Chlamydophila pneumoniae (I). (1) Lesión de los vasos por diferentes causas. (2) Esta lesión incrementa la síntesis de proteína quimiotáctica 1 de monocito y moléculas de adhesión que fijan leucocitos infectados; actividad procoagulante de lipoproteínas tisulares, con trombosis y adhesión de plaquetas. La célula endotelial se infecta por C. pneumoniae intramonocítica. (3) Los monocitos migran al subendotelio, liberan citocinas proinflamatorias y expresan la molécula CD14. El interferón 7 aumenta la dioxigenasa intramonocítica que oxida el triptófano hasta quineurina. (4) Se infectan las células musculares lisas y (5) los macrófagos.
. (1) La quineurina reduce la salida de C. pneumoniae y aumenta la expresión de la proteína de choque térmico 60 (HSP 60). (2) Los macrófagos infectados engloban las lipoproteínas de baja densidad (LDL) y se transforman en células grasas. (3) Las plaquetas agregadas liberan un factor de crecimiento celular que hace proliferar un músculo liso indiferenciado que libera colágeno, elastina y proteoglucanos con formación de tejido fibroso. (4) Se forma la placa madura con un core de lípidos y colesterol rodeada de una capa fibrosa.")
Patogenia de la arteriosclerosis y su relación con la infección por Chlamydophila pneumoniae (II). (1) La quineurina reduce la salida de C. pneumoniae y aumenta la expresión de la proteína de choque térmico 60 (HSP 60). (2) Los macrófagos infectados engloban las lipoproteínas de baja densidad (LDL) y se transforman en células grasas. (3) Las plaquetas agregadas liberan un factor de crecimiento celular que hace proliferar un músculo liso indiferenciado que libera colágeno, elastina y proteoglucanos con formación de tejido fibroso. (4) Se forma la placa madura con un core de lípidos y colesterol rodeada de una capa fibrosa.
La célula endotelial infectada incrementa la síntesis, por un lado, de la proteína quimiotáctica-1 del monocito y también de moléculas de adhesión (selectina E, molécula de adhesión intercelular-1, y molécula de adhesión vascular-1, que fijan nuevos leucocitos, los cuales pueden estar infectados) y, por otro lado, de factores tisulares procoagulantes, lipoproteínas tisulares, con trombosis y adhesión de plaquetas57.
Los monocitos, que no son susceptibles al ciclo replicativo completo de la bacteria, se activan, migran al subendotelio, liberan citocinas proinflamatorias (factor de necrosis tumoral [TNF], IL-1, 6 y 8, factor nuclear kappa B) y expresan la molécula CD1458. El IFN-γ, de los LTh1 sensibilizados59, aumenta la dioxigenasa intramonocítica de las células endoteliales y mononucleares, y oxida el triptófano hasta N-formilquineurina y quineurina (fig. 4). Esta sustancia reduce, según hemos visto en el ciclo replicativo y la patogenia de la infección, la salida de los cuerpos elementales (lo que explican los falsos negativos del cultivo de placas de ateroma), aumenta la persistencia intracelular porque surgen las formas crípticas denominadas CPE, reduce la expresión de la MOMP y aumenta la de la HSP 6060,61. Así, se ha visto que si no hay arteriosclerosis, no se observa la HSP 60 en la pared arterial. Ésta estimula la síntesis de autoanticuerpos y, de nuevo, a los macrófagos, produciendo más citocinas y metaloproteinasa que degradan el colágeno62. De esta manera, se establecería una infección crónica con presencia de CPE en la que el antígeno más abundante es el lipopolisacárido63.
Kalayoglu y Byrne54 demostraron que los macrófagos infectados engloban más LDL, por acción del lipopolisacárido, transformándose en células grasas. Las plaquetas agregadas liberarían un factor de crecimiento celular que hace proliferar un músculo liso indiferenciado, que libera colágeno, elastina y proteoglucanos con formación de tejido fibroso. Finalmente, se forma la placa de ateroma madura, con un centro de lípidos y colesterol, rodeada de una capa fibrosa.
Acerca de la asociación entre la enfermedad arterial periférica (EAP) oclusiva y la infección por esta bacteria se han publicado más de 50 estudios64, caracterizados por un grupo de casos y controles y metodologías analíticas bien definidas, en los que se analizan, mediante alguna prueba independiente, esta relación. De éstos, se ha concluido que existe, de forma estadísticamente significativa, una asociación cuando se utilizan pruebas inmunohistoquímicas, PCR anidada en biopsias de arterias, estudios de PCR en muestras no arteriales, pruebas de ELISA y MIF para detectar valores altos de inmunoglobulina G (IgG) e IgA, y otros métodos directos de detección de la bacteria diferentes de los anteriores. Sin embargo, no se ha encontrado asociación cuando se ha empleado la PCR simple en biopsias arteriales, en las pruebas de MIF que detectan IgG en niveles bajos, o IgM, y en los estudios de ELISA para detectar IgM. Según lo anterior, el hallazgo de la asociación entre la EAP y la infección por C. pneumoniae depende del método empleado en estos trabajos. La mayoría establece una relación con una probabilidad variable, pero no se encuentra esta relación con los métodos de PCR menos sensibles y la presencia de IgM antibacteria, lo que parecería lógico, ya que la EAP hay que entenderla como una enfermedad crónica. En relación con la detección del ADN de C. pneumoniae, en la mayor parte de los estudios publicados sólo se informa de las regiones amplificadas, que han sido muy diferentes, pero no del método de extracción del ADN y de la sensibilidad de la prueba empleada. Esto puede justificar las discrepancias de los resultados obtenidos en los estudios, ya que las metodologías han sido muy diferentes.
Aunque la asociación arteriosclerosis-Chlamydophila es aparente en muchos estudios, no implicaría que exista una relación causal, ya que faltan, entre otros, estudios que empleando un análisis de los marcadores de la inflamación justifiquen la relación patogénica causa-efecto.
Finalmente, hay que señalar que otros agentes infecciosos pueden estar involucrados en la aparición de la arteriosclerosis65, pues, en general, las infecciones crónicas pueden ejercer un efecto proaterogénico al actuar a nivel sistémico o de forma local sobre la pared vascular66.
En conclusión, aún nos encontramos en estadios muy iniciales en el conocimiento de la proteómica de C. pneumoniae. Su estudio ayudará decisivamente a conocer los mecanismos patogénicos finales por los que ocasiona la enfermedad en el hombre y, por tanto, a saber si esta bacteria desempeña un papel en enfermedades como la aterosclerosis.
Este estudio fue financiado por el Proyecto de Investigación CTS-138 de la Consejería de Innovación, Ciencia y Empresa de la Junta de Andalucía (Descripción de inmunógenos de Chlamydia pneumoniae reconocidos por los sueros de sujetos con enfermedad arterial periférica. Relaciones clínicas).

