



La trimetilaminuria primaria o síndrome de olor a pescado es un error innato del metabolismo, debido al defecto en la oxidación hepática de la trimetilamina (TMA) a trimetilamina N-óxido (TMANO). La TMA procede del metabolismo de precursores dietéticos como colina, carnitina y TMANO. Su excreción en cantidades anormales en la orina, el sudor, el aire espirado y el resto de secreciones corporales despide un fuerte olor que recuerda al pescado podrido. Los pacientes afectos generalmente desarrollan graves problemas psicosociales. Presentamos un caso de trimetilaminuria primaria, con el fin de contribuir al conocimiento de este trastorno.
Primary trimethylaminuria, or fish odor syndrome, is a congenital metabolic disorder characterized by a failure in the hepatic trimethylamine (TMA) oxidation route to trimethylamine N-oxide (TMANO). TMA is mostly derived from dietary precursors such as choline, carnitine and TMANO. The presence of abnormal amounts of TMA in the urine, sweat, exhaled air and other body secretions confers a very unpleasant body odor resembling that of decaying fish. As a consequence, patients can suffer from serious psychosocial sequelae. We present a case of primary trimethylaminuria with the aim of raising awareness about this condition.
La trimetilaminuria o síndrome de olor a pescado es una metabolopatía poco frecuente que se caracteriza por un defecto del sistema enzimático hepático flavinmonooxigenasa 3 (FMO3). En individuos sanos, la enzima hepática reoxida la trimetilamina (TMA) en el compuesto inodoro trimetilamina N-óxido (TMANO).
La excreción masiva de TMA en la orina, el sudor, el aire espirado y el resto de las secreciones corporales no es tóxica, pero confiere un intenso olor corporal a pescado. Las consecuencias clínicas de esta afección son de naturaleza psicosocial, fruto del rechazo social que sufren los pacientes y la falta de explicación a sus síntomas. En muchos se desarrollan trastornos de la personalidad, obsesión por la higiene corporal, cuadros de ansiedad y síndromes depresivos graves1. Los primeros síntomas aparecen en la infancia, pero probablemente el desconocimiento médico, junto con el desconocimiento del paciente, que en muchas ocasiones se muestra reacio a consultar, es la principal causa de la tardanza diagnóstica.
La confirmación diagnóstica es sencilla, se realiza mediante cuantificación en orina de TMA y TMANO2. Actualmente no existe tratamiento etiológico, pero la eliminación de la dieta de forma controlada de alimentos ricos en colina y TMANO, que son los principales precursores dietéticos de la TMA, reduce en gran medida los síntomas y los pacientes mejoran considerablemente su calidad de vida.
CASO CLÍNICOMujer de 30 años que sufría desde la infancia mal olor corporal, que describía como semejante al olor del pescado. Este síntoma se exacerbaba con la ingestión de pescado marino, huevos y legumbres. Había consultado en varias ocasiones sin obtener respuesta a su problema. Cuando acudió a nuestro centro se había documentado a través de internet y conocía la existencia de este trastorno. La paciente carecía de antecedentes personales de interés. La exploración física fue normal y se descartó trastornos hepáticos y renales.
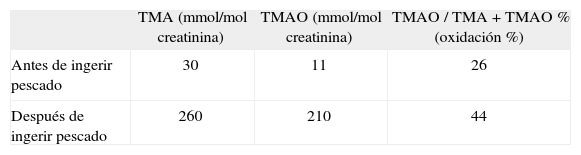
Con la sospecha clínica de un caso de trimetilaminuria primaria, nos pusimos en contacto con el Laboratorio de Pediatría y Neurología de la Universidad Radboud en Nijmegen (Países Bajos), centro de referencia en Europa para la trimetilaminuria, donde se realizó estudio bioquímico de la orina. Se remitieron dos muestras de orina, antes y tras 12h de haber ingerido 300mg de pescado marino fresco (test de sobrecarga) y se cuantificó la concentración de TMA y TMANO mediante espectroscopia de resonancia magnética. La concentración de TMA en la muestra de orina basal fue de 30 |imol/mmol de creatinina (normal: < 5μmol/mmol de creatinina) junto con concentraciones de TMANO indetectables, lo cual es diagnóstico de trimetilaminuria. Tras la sobrecarga de pescado, se objetivó un incremento de las cifras de TMA a 260μmol/mmol de creatinina y de TMANO a 210μmol/mmol de creatinina (tabla 1).
Resultados del estudio bioquímico
| TMA (mmol/mol creatinina) | TMAO (mmol/mol creatinina) | TMAO / TMA + TMAO % (oxidación %) | |
| Antes de ingerir pescado | 30 | 11 | 26 |
| Después de ingerir pescado | 260 | 210 | 44 |
TMA: trimetilamina; TMANO: trimetilamina N-óxido. Valores de referencia antes de ingerir pescado: TMA < 5; TMANO, 10–500; valores de referencia de TMAO / TMA + TMAO%, 95-98%.
Tras la confirmación bioquímica, se solicitó estudio genético, que mostró una mutación no descrita previamente. Se analizó la región codificante del gen FMO3 mediante electroforesis en gradiente desnaturalizante y las anomalías encontradas se caracterizaron por secuenciación fluorescente de ADN. La paciente resultó ser homocigota para la variante T307P (c.919A>C) en el exón 7 del gen FMO3.
De acuerdo con las preferencias de la paciente, se diseñó una dieta controlada en colina, normocalórica, con un aporte adecuado de hidratos de carbono, grasas y proteínas de alto valor biológico (0,8g/kg). Se añadió un complejo vitamínico rico en acido fólico y riboflavina. Se la reevaluó a los 3 meses, con clara mejoría de los síntomas.
DISCUSIÓNLa trimetilaminuria es un ejemplo excelente de cómo la genética puede interaccionar negativamente con la dieta. Esta rara condición producida por un defecto en el metabolismo de la TMA fue descrita por primera vez por Humbert et al3 en 1970. La TMA es una amina terciaria muy volátil, que confiere un desagradable olor a pescado a la orina, el sudor y demás secreciones corporales. La TMA procede del metabolismo intestinal de alimentos ricos en colina, lecitina, carnitina y TMANO. En condiciones normales, el 95% de la TMA sufre una N-oxidación en el hígado mediada por la enzima FMO3, que la transforma en TMANO, compuesto inodoro que se elimina por la orina4 (fig. 1). Aunque se reconocen variaciones étnicas en la capacidad oxidativa de la enzima5, los pacientes afectos de trimetilaminuria no degradan la TMA en TMANO y se produce una excreción masiva de trimetilamina no oxidada. La enzima FMO3 además interviene en el metabolismo del nitrógeno y en la desintoxicación de otras aminas endógenas, tiramina, nicotina y fármacos como los antidepresivos tricíclicos y la ranitidina4.

La incidencia estimada de esta afección es de 1 caso cada 40.000 personas, ya que el 1% de la población general podría ser portadora de este defecto4-6. En España se han publicado únicamente 3 casos7-9 (búsqueda bibliográfica realizada sin limitar fecha de inicio hasta enero de 2009, utilizando los términos en inglés y castellano: "trimetilaminuria", "trimetilamina", "síndrome de olor a pescado", "flavinmonooxigenasa 3"). A la vista de estos datos, es una alteración claramente infradiagnosticada.
La trimetilaminuria primaria es un trastorno autosómico recesivo debido a la mutación del gen FMO3 localizado en el brazo largo del cromosoma 110,11. Dicho gen es altamente polimórfico; se han descrito al menos 40 mutaciones, solas o en combinación, que se asocian con una mayor o menor actividad de la enzima, lo que hace posible que los individuos que padecen la enfermedad desarrollen formas más o menos graves. Los casos más severos de trimetilaminuria presentan las mutaciones P153L y E305X12.
Las formas secundarias de trimetilaminuria se deben a lesiones renales o hepáticas13,14. Además se han descrito formas transitorias relacionadas con factores que modifican la capacidad oxidativa de la enzima: infecciones virales, inmadurez del sistema oxidativo (niños prematuros), inhibidores enzimáticos, exceso de precursores dietéticos de TMA4 (dieta enriquecida en colina, problemas renales o hepáticos, alteración de la microflora intestinal, incremento de la absorción intestinal) y factores hormonales1,15 (episodios de trimetilaminuria durante la menstruación). Estas formas intermedias o transitorias suelen aparecer en individuos heterocigotos6.
Las consecuencias clínicas están definidas en el nombre coloquial de la enfermedad: "síndrome de olor a pescado". Los primeros síntomas de la trimetilaminuria primaria aparecen en la infancia al introducir alimentos ricos en colina y TMANO. Los padres suelen referir que su hijo "no huele a bebé". El olor se exacerba en situaciones de sudoración excesiva como la fiebre y el ejercicio intenso, durante la menstruación y la toma de anticonceptivos orales15. Todos los autores coinciden en que el mal olor corporal produce un impacto demoledor en la vida personal, social y laboral de los pacientes— por lo que muchos desarrollan hábitos compulsivos de higiene para disimular su olor— junto con trastornos de tipo psicosocial, ansiedad, síndromes depresivos e incluso intentos de suicidio1,16. Se desconoce si estos síntomas se pueden atribuir al metabolismo patológico de la tiramina y otros neurotransmisores que son sustratos de la enzima FMO34. Por otra parte, la alteración del metabolismo del nitrógeno se asocia a hipertensión arterial e incremento del riesgo cardiovascular2.
En la mayoría de los casos el diagnóstico se retrasa una media de 10 años8 debido al desconocimiento médico de este trastorno y a que los pacientes aplazan la consulta por miedo a que se les atribuya falta de higiene. Aunque es un defecto autosómico recesivo, no ligado al sexo, se diagnostica más en mujeres, lo que podría deberse a la influencia de los factores hormonales o que las mujeres toleren peor los síntomas. El diagnóstico diferencial incluye la vaginosis bacteriana, infecciones urinarias, dimetilglicinuria y enfermedades hepáticas o renales17.
La confirmación diagnóstica se obtiene mediante la determinación bioquímica en orina del cociente TMANO / (TMA + TMANO) en orina2. Este cociente está drásticamente disminuido en los pacientes homocigotos, pero requiere un test de sobrecarga (600mg de TMA) para distinguir a los individuos sanos de los pacientes heterocigotos6.
El estudio del genotipo molecular permite detectar la mutación asociada a esta enfermedad. En el caso presentado la mutación T307P (c.919A>C) produce la sustitución del aminoácido arginina por glicina en la estructura primaria de la proteína FMO3. Se trata de una mutación no descrita previamente.
A día de hoy no existe tratamiento etiológico, pero la eliminación controlada de la dieta de alimentos ricos en colina y TMANO minimiza el mal olor y los pacientes mejoran.
La colina se encuentra fundamentalmente en huevos, vísceras, carnes magras, salmón y bacalao, guisantes, judías, espinacas, coles, en algunas legumbres como las alubias, los frutos secos y los productos de comida rápida (fastfood) como la lasaña, hamburguesas y pizza. La TMANO proviene del pescado marino y los crustáceos.
Los requerimientos de colina en individuos sanos son 550mg/día (varones) y 425mg/día (mujeres)18. La colina plasmática y la fosfatidilcolina forman parte de la membrana celular, son necesarias para su integridad estructural y participan en la señalización transmembrana, la neurotransmisión colinérgica y el transporte y el metabolismo de los lípidos19. Las dietas deficientes en colina deben ajustarse individualmente, ya que una dieta muy restrictiva puede producir hígado graso, retraso del crecimiento, alteraciones óseas y disfunción renal20. No se recomienda la restricción de colina en embarazadas ni en niños en edad de crecimiento. Las cifras mínimas de colina deben ser 100mg/día18 (dieta muy restrictiva); estas dietas deben ser realizadas por un nutricionista, ya que requieren la inclusión de proteínas de alto valor biológico (clara de huevo o soja), cantidades moderadas de grasa (30%) como fuente de energía y fruta y verdura en cantidad adecuada. Además los pacientes deben ser suplementados con complejos vitamínicos que contengan el 100% de las CDR (cantidad diaria recomendada) para vitaminas y minerales, especialmente ácido fólico y riboflavina, ya que el déficit de colina conlleva el uso de folato en la metilación de la homocisteína a metionina y, por lo tanto, la depleción de los depósitos de ácido fólico21.
Como tratamiento coadyuvante, se ha propuesto el uso de ciclos cortos de neomicina, metronidazol (500mg/12h durante 10 días) y lactulosa para disminuir la producción de TMA por la flora intestinal2,22. También ha resultado efectivo el uso de resinas de intercambio iónico, carbón activado (1,5g/día durante 10 días) y cobre-clorofilina23 (180mg/día durante 21 días). Se aconseja el uso de estas medidas de forma periódica y en momentos de mayor estrés, infecciones o durante la menstruación. Además se deben evitar los fármacos que puedan interferir en el metabolismo hepático y se recomienda el uso de jabones con pH 5,5-6,5 para eliminar la TMA de la piel24. Asimismo existen grupos de apoyo como la Trimethylaminuria Foundation (trimeth411@aol.com) que proporciona información a los pacientes.
Las estrategias propuestas para el futuro incluyen modificar los genes mutados de la FMO3 y colonizar el intestino con microorganismos mutados genéticamente con FMO3 humana4, aunque habrá que valorar el coste-beneficio de estas terapias.
Con la presentación de este caso, hemos querido contribuir al conocimiento de este desafortunado trastorno metabólico. Mantener un alto grado de sospecha diagnóstica permitirá una confirmación precoz y una temprana instauración de la dieta, condición vital para estos pacientes.
