



Se revisan: la hiperplasia suprarrenal macronodular bilateral independiente de corticotropina (ACTH), caracterizada por la expresión aberrante de receptores en la corteza adrenal. Como expresión ectópica o hay una expresión amplificada (expresión eutópica). La hiperplasia adrenal micronodular, que cursa con pequeños nódulos pigmentados en la adrenal con atrofia del tejido internodular. Estos nódulos pueden no ser vistos en las pruebas de imagen.
El síndrome de Cushing subclínico, introducido en 1981, debe aplicarse a los pacientes que tienen adenomas adrenales clínicamente no funcionantes, pero con secreción de cortisol autónoma, aunque insuficiente para producir la aparición de un cuadro florido. Conviene distinguirlo del síndrome de Cushing preclínico, dado que no se trata necesariamente de un estado previo a la clínica de hipercortisolismo.
El síndrome de Cushing es poco frecuente en niños y adolescentes. De la incidencia general de la enfermedad, sólo el 10% de los casos son diagnosticados en esas edades. La causa más frecuente de síndrome de Cushing endógeno en niños mayores de 7 años es la enfermedad de Cushing dependiente de ACTH (85%).
La combinación síndrome de Cushing y embarazo es muy poco frecuente. Su rareza se debe que el hipercortisolismo habitualmente cursa con amenorrea, oligomenorrea e infertilidad por inhibición de la secreción de gonadotropinas. Se han descrito 136 gestaciones en 122 mujeres, con una edad gestacional al diagnóstico de 18,4 ± 1 semanas.
The present article reviews: Corticotrophin (ACTH) independent bilateral macronodular adrenal hyperplasia, which is characterized by aberrant adrenal receptors due to either ectopic expression or to overexpression (eutopic expression). Micronodular adrenal hyperplasia, which provokes small pigmented nodules in the adrenal gland with atrophy of the internodal tissue. These nodules may not be visible on imaging tests.
The term subclinical Cushing's syndrome, coined in 1981, should be used in patients with clinically non-functioning adrenal adenomas but who show autonomous cortisol production that is insufficient to generate overt Cushing's syndrome. This entity must be distinguished from preclinical Cushing's syndrome, given that the subclinical form does not necessarily herald the development of symptoms of hypercortisolism.
Cushing's syndrome is uncommon in children and adolescents. Regarding the general incidence of the disease, only 10% of cases are diagnosed in this age group. The most common cause of endogenous Cushing's disease in children older than 7 years is ACTH-dependent Cushing's disease (85%).
The association of Cushing's syndrome and pregnancy is highly uncommon, since hypercortisolism usually causes amenorrhea, oligomenorrhea and infertility due to inhibition of gonadotropin secretion. One hundred thirty-six pregnancies have been described in 122 women, with a gestational age at diagnosis of 18.4 ± 1 weeks.
La hiperplasia nodular es, dentro de las formas de síndrome de Cushing independiente de corticotropina (ACTH), una rara causa de hipercortisolismo.
Incluye tres formas de hiperplasia adrenal perfectamente diferenciadas: la hiperplasia macronodular, la enfemedad nodular pigmentaria (primaria o incluida en el complejo de Carney) y la hiperplasia incluida en el síndrome de McCune-Albright. Cada una de ellas afecta a menos del 2% de los pacientes con síndrome de Cushing, y la relación por sexos es 1:1.
Hiperplasia suprarrenal macronodular bilateral independiente de ACTHSe caracteriza por la expresión aberrante de receptores en la corteza adrenal. Estos receptores normalmente no están (expresión ectópica) o hay una expresión amplificada de receptores habitualmente presentes (expresión eutópica)1.
El síndrome clínico se evidencia durante la quinta o sexta década de la vida. La mayoría de los casos son esporádicos, pero se han descrito algunos casos familiares. Pueden diagnosticarse en fase de síndrome de Cushing subclínico, a partir de un hallazgo incidental radiológico2,3. Así, un 10-15% de los incidentalomas adrenales bilaterales se consideran que son formas subclínicas de la enfermedad, con una concentración de ACTH no suprimida completamente4.
En algunos pacientes, se ha descrito secreción de cortisol mediada por receptores funcionales de membrana para GIP (péptido inhibidor gástrico), lo que da lugar a incrementos transitorios de cortisol tras las comidas, con concentraciones normales en los periodos de ayuno. En pacientes con expresión de receptores V1-vasopresina, el incremento de cortisol ocurre en respuesta al cambio de postura al ortostatismo o a otros estímulos de la vasopresina endógena. La expresión aberrante de receptores betaadrenérgicos se identificó en pacientes con elevaciones de cortisol que coinciden con aumentos de catecolaminas endógenas (en respuesta a ortostatismo, hipoglucemia, ejercicio, administración de isoproterenol etc.). La de receptores LH/ hCG se ha descrito en varios casos de Cushing que se desarrolla durante la gestación o en la fase posmenopáusica. Se ha publicado en varios pacientes el estímulo del cortisol tras administración de cisaprida y metoclopramida (agonistas serotoninérgicos 5-HT4) o en combinación con otros posibles estimulantes de receptores aberrantes. Estudios in vitro han demostrado la expresión de otros receptores como interleucina 1, leptina y, posiblemente, por otros ligandos no reconocidos hasta ahora.
Las causas de la anormal expresión de estos receptores siguen siendo desconocidas. Los receptores aberrantes también pueden estar en adenomas unilaterales, pero mucho menos frecuentemente que en la hiperplasia bilateral. Además, se han descrito casos de pacientes con enfermedad dependiente de ACTH en los que se ha demostrado expresión de receptores de GIP en la corteza adrenal.
La mayoría de estos receptores ectópicos han sido demostrados en estudios de incubación in vitro, hibridación in situ, y mediante RT-PCR. Se ha demostrado que el tejido hiperplásico puede expresar simultáneamente múltiples receptores de membrana para factores paracrinos-autocrinos circulantes e intraadrenales. De hecho, se ha visto que las células de estas hiperplasias producen sustancias de efecto corticotropo por ellas mismas, evidenciando la regulación autocrina-paracrina de la secreción tisular de cortisol5.
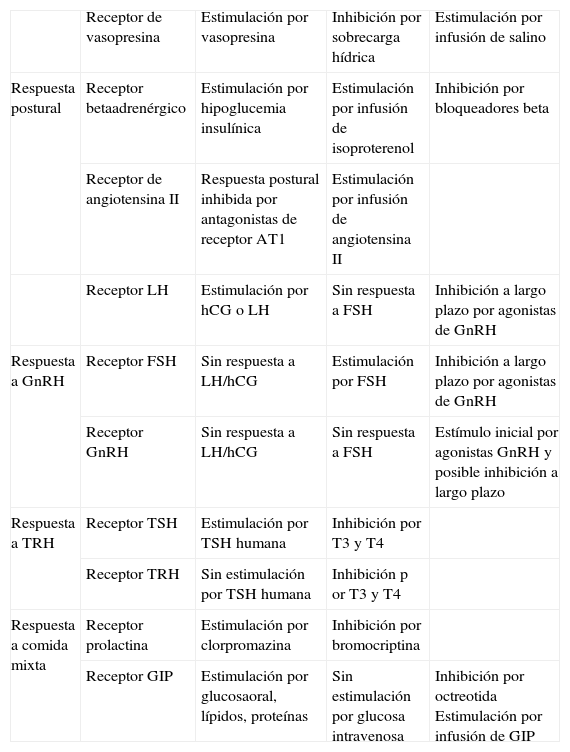
Se ha desarrollado un protocolo de cribado para detectar estos receptores6. Consiste en la determinación de cortisol durante varias pruebas con estímulos que transitoriamente modulan las concentraciones de ligandos para receptores potencialmente aberrantes. Incluye medidas de ACTH, cortisol y otros esteroides y hormonas a intervalos de 30–60min durante 2 o 3h. En el primer día las pruebas se realizan después de una noche de ayuno y en posición supina, seguido por un periodo de 2h de deambulación, para evaluar la modulación por angiotensina II, vasopresina, catecolaminas, pruebas, etc. Después se sigue de una comida mixta estándar para evaluar la respuesta a las hormonas gastrointestinales (por ejemplo, GIP). A continuación se administran 250μg de ACTH 1–24 intravenoso (i.v.), que sirve como prueba de referencia de la capacidad de respuesta cortical. En el segundo día de estudio se administran 100μg de gonadoliberina (LHRH) i.v. (modulación por folitropina, luteotropina, LHRH) seguidos por 200μg de TRH i.v. (modulación por tirotropina, prolactina, TRH). El tercer día de estudio consiste en administrar 1mg de glucagón i.v., 10 U AVP intramuscular y 10mg de metoclopramida o cisaprida oral (agonistas de receptores 5-HT4).
Para valorar el resultado, un cambio de menos del 25% de cortisol se define como sin respuesta, un cambio del 25-49% se define como respuesta parcial y un cambio del 50% o mayor, como respuesta positiva.
Si se obtiene respuesta parcial o positiva, la prueba se repite para confirmación y determinación de si otros esteroides, como aldosterona, sulfato de dehidroepiandrosterona, testosterona y estradiol también se modifican. A la vez, se miden también fluctuaciones de otras hormonas potencialmente implicadas como ligandos (catecolaminas, vasopresina, renina/angiotensina II y pruebas durante el test de la postura). Si una respuesta a una prueba es prolongada, puede enmascarar la evaluación de la siguiente prueba, por lo que deben repetirse separadamente.
Después de las pruebas de cribado, pueden realizarse otras pruebas (tabla 1) para confirmar las respuestas o aclarar cuál es la hormona implicada.
Protocolo de caracterización de receptores hormonales suprarrenales anormales cuando una prueba ha sido positiva en el cribado inicial
| Receptor de vasopresina | Estimulación por vasopresina | Inhibición por sobrecarga hídrica | Estimulación por infusión de salino | |
| Respuesta postural | Receptor betaadrenérgico | Estimulación por hipoglucemia insulínica | Estimulación por infusión de isoproterenol | Inhibición por bloqueadores beta |
| Receptor de angiotensina II | Respuesta postural inhibida por antagonistas de receptor AT1 | Estimulación por infusión de angiotensina II | ||
| Receptor LH | Estimulación por hCG o LH | Sin respuesta a FSH | Inhibición a largo plazo por agonistas de GnRH | |
| Respuesta a GnRH | Receptor FSH | Sin respuesta a LH/hCG | Estimulación por FSH | Inhibición a largo plazo por agonistas de GnRH |
| Receptor GnRH | Sin respuesta a LH/hCG | Sin respuesta a FSH | Estímulo inicial por agonistas GnRH y posible inhibición a largo plazo | |
| Respuesta a TRH | Receptor TSH | Estimulación por TSH humana | Inhibición por T3 y T4 | |
| Receptor TRH | Sin estimulación por TSH humana | Inhibición p or T3 y T4 | ||
| Respuesta a comida mixta | Receptor prolactina | Estimulación por clorpromazina | Inhibición por bromocriptina | |
| Receptor GIP | Estimulación por glucosaoral, lípidos, proteínas | Sin estimulación por glucosa intravenosa | Inhibición por octreotida Estimulación por infusión de GIP |
AT1: angiotensina 1; FSH: folitropina; GIP: péptido inhibidor gástrico; GnRH: gonadoliberina; LH: luteotropina; T3: triyodotironina; T4: tiroxina.
En los trabajos publicados con aplicación sistemática de este protocolo de cribado, todos los casos de hiperplasia han tenido respuesta positiva al menos a una prueba, aparte de la correspondiente respuesta a ACTH exógena. Se ha descrito respuesta en algún caso de adenoma suprarrenal y en ninguno de carcinoma.
Una limitación de esta prueba es que no permite estudiar otros receptores de membrana acoplados a proteína G, tales como paratirina, calcitonina, acetilcolina, dopamina, opiáceos, prostaglandina.
Hiperplasia primaria adrenal nodular pigmentadaTambién conocida como "hiperplasia adrenal micronodular", cursa con pequeños nódulos pigmentados en la adrenal con atrofia del tejido internodular. Estos nódulos pueden no visualizarse en las pruebas de imagen, y el tamaño de las glándulas es normal. El diagnóstico, además, está dificultado porque la clínica puede ser leve y cíclica. Puede presentarse de forma aislada en el 50% de los casos o incluido en el complejo de Carney (un tipo de síndrome de neoplasia endocrina múltiple) que, en esta enfermedad, es la manifestación endocrina más frecuente (otros datos clínicos son lentigos, mixomas, schwannomas, etc.). Se presenta al final de la infancia o la adolescencia; hay descritas varias series de pacientes pediátricos con esta rara enfermedad7.
De herencia autosómica dominante, se debe a mutaciones germinales inactivantes de la subunidad 1-α de la proteincinasa A (PRKAR1A), localizada en el cromosoma 17q22-24. Están en el 45% de los pacientes con complejo de Carney y en las formas esporádicas de esta hiperplasia. En pacientes con sospecha diagnóstica debe realizarse esta prueba genética.
Estos pacientes muestran una respuesta paradójica del cortisol a la supresión con dexametasona, con un incremento mayor del 50% del cortisol libre urinario (no ocurre en las otras formas de hiperplasia), en relación con un aumento de la expresión del receptor de glucocorticoides en los nódulos, confirmado en estudios in vitro8.
La alta expresión de sinaptofisina en los nódulos indica el fenotipo neuroendocrino de estas células3.
Síndrome McCune-AlbrightLa hiperplasia es un hallazgo raro y no siempre en relación con el síndrome de Cushing. Las mutaciones activadoras en el gen GNAS1 (mutaciones somáticas poscigóticas), llamadas gsp llevan a un aumento de la esteroidogénesis en los nódulos que tienen la mutación. Estas mutaciones están dispersas, con un patrón tipo mosaico, en los distintos tejidos del organismo. Ocurren durante el desarrollo embriológico temprano, que lleva a una dispersión en la distribución de las células que presentan la mutación, lo que conlleva un amplio espectro de fenotipos9. La enfermedad puede presentarse en las primeras semanas de vida.
TratamientoEn general, la adrenalectomía bilateral es el tratamiento de elección. Algunos autores plantean realizar adrenalectomía unilateral, con buenos resultados iniciales, aunque con incremento posterior de la glándula contralateral, que hace necesaria la segunda adrenalectomía. Está la posibilidad de realizar la adrenalectomía por vía laparoscópica, aunque se alarga en estos casos el tiempo quirúrgico, por lo que se puede plantear la opción de realizar la primera adrenalectomía por vía laparoscópica y la segunda por laparotomía abierta10.
En la forma subclínica no está establecido cuál debe ser el tratamiento, que depende de los síntomas; se aconseja, en la mayoría de los casos, seguimiento anual.
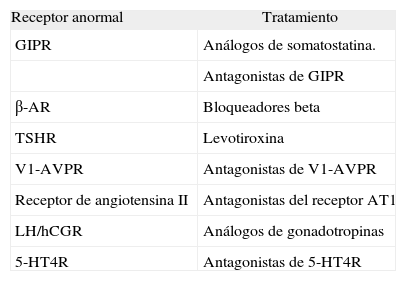
La caracterización de estos receptores aberrantes abre la posibilidad de una aproximación farmacológica como alternativa a la adrenalectomía (tabla 2).
Posibles tratamientos farmacológicos para receptores anormales en tumores adrenocorticales
| Receptor anormal | Tratamiento |
| GIPR | Análogos de somatostatina. |
| Antagonistas de GIPR | |
| β-AR | Bloqueadores beta |
| TSHR | Levotiroxina |
| V1-AVPR | Antagonistas de V1-AVPR |
| Receptor de angiotensina II | Antagonistas del receptor AT1 |
| LH/hCGR | Análogos de gonadotropinas |
| 5-HT4R | Antagonistas de 5-HT4R |
GIPR: receptor del péptido inhibidor gástrico; AT1: angiotensina 1.
Hay experiencias iniciales de tratamiento con leuprolida, propanolol y análogos de somatostatina, pero es necesario obtener mayor información y experiencia antes de considerar el lugar de este tipo de tratamiento en el manejo de la hiperplasia adrenal macronodular.
Es posible que, a pesar de la inhibición completa de los receptores aberrantes, la regresión tumoral no ocurra porque se den otros procesos genéticos que, a su vez, puedan inducir mutaciones que condicionen un aumento de función3.
SÍNDROME DE CUSHING SUBCLÍNICOHa adquirido especial interés en los últimos años por el progresivo incremento del hallazgo de incidentalomas adrenales, unido al interés creciente por el estudio y el tratamiento de enfermedades en fase subclínica.
El término de síndrome de Cushing subclínico, introducido en 1981, debe aplicarse a los pacientes que tienen adenomas adrenales clínicamente no funcionantes, pero con secreción de cortisol autónoma, aunque insuficiente para producir la aparición de un cuadro florido11. Conviene distinguirlo del término Cushing preclínico dado que no se trata necesariamente de un estado previo a la clínica de un hipercortisolismo ni de una fase evolutiva de éste.
Deben cumplirse dos criterios para su diagnóstico: el paciente no debe presentar fenotipo cushingoide, y debe tener una masa adrenal descubierta incidentalmente. Aunque podría aplicarse también al incidentaloma hipofisario, cuando se emplea este término nos estaremos refiriendo exclusivamente al adrenal. Se ha publicado algún caso debido a carcinoma adrenal o mielolipoma que, tras la cirugía, se demuestra que se trataban de tumores mixtos.
La prevalencia oscila en la literatura entre el 5 y el 20% de los pacientes con incidentalomas adrenales, según las pruebas diagnósticas empleadas y los criterios aplicados.
En general, cuanto mayor es el número de alteraciones bioquímicas al diagnóstico, hay mayor probabilidad de aumento de tamaño del adenoma o desarrollo de adenoma bilateral12.
DiagnósticoSegún las series publicadas, en el estudio hormonal de los incidentalomas adrenales, se encuentran alteraciones hasta en el 28% de los casos; las más frecuentes son una disminución de las concentraciones de DHEAS y ACTH13. No es fácil el diagnóstico porque el grado de hipercortisolismo sólo excede ligeramente las concentraciones fisiológicas, con lo que se evidencia un amplio espectro de variabilidad entre el adenoma no funcionante y la producción autónoma de cortisol. De hecho, puede considerarse más una alteración cualitativa que cuantitativa de la secreción de cortisol, lo que constituye una condición o entidad muy heterogénea.
El hallazgo más frecuente en los incidentalomas adrenales es la determinación de concentraciones bajas de DHEAS. Según algunos autores reflejaría la supresión de la ACTH por la secreción autónoma de cortisol, aunque hay una mala correlación entre esta prueba y otros resultados. Además, hay que tener en cuenta que las concentraciones de DHEAS disminuyen con la edad.
La pérdida del ritmo circadiano es también un hallazgo frecuente, con elevación del cortisol nocturno; este dato se considera el primer marcador del cuadro. Las concentraciones de cortisol basales suelen ser normales. En la mayor parte de los pacientes el cortisol libre urinario es normal. Las concentraciones de ACTH son bajas o indetectables. La respuesta de ACTH y cortisol al CRH puede estar anulada, aunque la interpretación de esta prueba puede ser confusa. En la gammagrafía con yodo-colesterol, estos adenomas muestran captación unilateral con ausencia de captación de la glándula contralateral. Varios estudios han correlacionado esta captación del adenoma con la supresión de ACTH, y concluyen que esta captación representa un signo precoz de esta autonomía funcional, que puede ocurrir con pruebas bioquímicas normales. Según otros autores, puede reflejar simplemente un aumento del tejido glandular. No obstante, la gammagrafía no debe utilizarse sistemáticamente en estos pacientes11. Por otra parte, se ha demostrado que en estos adenomas hay un aumento de la captación de fluorodesoxiglucosa en la tomografía por emisión de positrones, a diferencia de los adenomas no funcionantes, lo que también indicaría una actividad metabólica y hormonal incrementada14.
Los algoritmos diagnósticos varían entre los distintos centros, pero la mayoría coincide en que deberían demostrarse alteraciones en 2 de las pruebas para llegar a este diagnóstico; hay varias posibles combinaciones cuando se estudia el eje hipófiso-adrenal en detalle. Por lo tanto, no hay acuerdo sobre la estrategia diagnóstica que debe seguirse para detectar esta autonomía adrenal cortical, ni a partir de qué grado de cortisolemia está aumentada la morbilidad.
Hay escasa información sobre los efectos de este síndrome a largo plazo, aunque es lógico pensar que estos pacientes tendrán las complicaciones del síndrome de Cushing en algún grado. Se ha confirmado en distintos estudios una mayor incidencia de factores de riesgo cardiovascular y síndrome metabólico: alteración del metabolismo hidrocarbonado (del 20 al 75% de los casos), hipertensión arterial (30-100%), resistencia insulínica, obesidad (35-50%), dislipemia (50%), elevación del fibrinógeno12. Hay evidencia de que este leve hipercortisolismo es insuficiente para causar la alteración fenotípica típica del síndrome de Cushing, pero es suficiente para desarrollar resistencia insulínica y sus consecuencias clínicas.
Por otra parte, la prevalencia del hipercortisolismo subclínico en pacientes con diabetes mellitus tipo 2 está aumentada, que es 4–5 veces más frecuente, independientemente de la coexistencia de obesidad e hipertensión arterial15.
También hay datos contradictorios sobre las consecuencias en la densidad mineral ósea, pero no parece haber diferencias significativas respecto a individuos normales de iguales edad y sexo16.
Los estudios que evalúan el riesgo de progresión desde la condición subclínica a síndrome de Cushing clínico demuestran que esta evolución ocurre raramente12. Masas de 3cm o mayores es más probable que desarrollen hiperfunción asintomática que tumores de menor tamaño, y el riesgo parece estacionarse o disminuir después de 3–4 años de seguimiento. En algunos pacientes, se objetiva normalización espontánea del hipercortisolismo subclínico, lo que indica que esta secreción puede ser variable a lo largo del tiempo. No obstante, estos datos son resultado de estudios retrospectivos, que incluyen muestras pequeñas, distintos tiempos y diferentes estrategias de seguimiento. Por ello, no hay acuerdo sobre las recomendaciones de pauta de seguimiento de estos pacientes, aunque la aproximación más aconsejable es la realización de la prueba de supresión con 1mg de dexametasona anualmente, o antes si está clínicamente indicado11.
Incluso se debate si el beneficio del diagnóstico justifica los costes que implican las pruebas a realizar en estos pacientes paucisintomáticos.
TratamientoDado que las complicaciones a largo plazo no son conocidas, el manejo de estos tumores es empírico11. La primera opción de tratamiento parece ser la adrenalectomía con cobertura perioperatoria y postoperatoria con glucocorticoides por el riesgo de hipoadrenalismo. El tratamiento deberá suspenderse tras la demostración de normalidad del eje hipofisario-adrenal. No se han establecido guías de seguimiento de pacientes no sometidos a adrenalectomía, y tampoco se ha evidenciado una mayor tasa de mortalidad en los pacientes no intervenidos17.
El efecto a largo plazo de este tratamiento no está claro, pero parece mejorar los parámetros clínicos y bioquímicos de riesgo cardiovascular18, sin modificación de los parámetros de metabolismo óseo. No hay estudios comparativos con otras posibles actuaciones terapéuticas dirigidas a este objetivo, como modificaciones de estilo de vida e intervenciones farmacológicas cuya utilidad está plenamente confirmada. Por ello, hasta que la relación riesgo-beneficio del tratamiento quirúrgico esté aclarada, los clínicos deberán ser quienes decidan la conveniencia de la cirugía en los pacientes que tienen Cushing subclínico con alteraciones potencialmente atribuibles al exceso subliminal de cortisol. Los pacientes no aptos para cirugía deberían incluirse en un programa de seguimiento para detectar manifestaciones del síndrome metabólico, y tratarlas sintomáticamente con fármacos de beneficio probado en la prevención de eventos cardiovasculares y de la desmineralización ósea.
SÍNDROME DE CUSHING EN EDAD PEDIÁTRICAEl síndrome de Cushing es poco frecuente en niños y adolescentes, lo que constituye un importante reto diagnóstico y terapéutico para los pediatras dedicados a la endocrinología. De la incidencia general de la enfermedad, sólo el 10% de los casos son diagnosticados en estas edades.
Respecto a la distribución por sexos, a diferencia de la preponderancia femenina de la enfermedad en población adulta, en la edad prepuberal es más frecuente en varones, con igual distribución durante la pubertad y predominio en las niñas en edades pospuberales. No se ha confirmado que este dato se acompañe de diferencias en la intensidad de la hipercortisolemia o en las concentraciones de ACTH al diagnóstico en varones respecto a niñas en la población infantil, como han apuntado algunos autores19.
La causa más frecuente de síndrome de Cushing endógeno en niños mayores de 7 años es la enfermedad de Cushing dependiente de ACTH (85%); el corticotropinoma es el segundo adenoma hipofisario más frecuente en niños. En el 90% de los casos se trata de microadenomas. Un 20% de los casos de enfermedad de Cushing dependiente de ACTH se producen por secreción de ACTH ectópica.
El 15% restante son de causa independiente de ACTH; en un 30% de los casos corresponden a adenomas y en un 70%, a carcinomas adrenales. La enfermedad micronodular o macronodular es muy poco frecuente20. En niños menores de 7 años, la causa más frecuente es la adrenal, principalmente, carcinomas adrenales.
ClínicaLa exposición prolongada al exceso de glucocorticoides causa fundamentalmente en niños retraso de crecimiento y excesivo aumento de peso. El fenotipo típico suele estar ausente: la obesidad tiende a ser generalizada más que específicamente centrípeta, con incremento de grasa visceral y subcutánea21.
El indicador más sensible del hipercortisolismo en niños es el retraso de crecimiento, que a menudo precede a otras manifestaciones. Una disminución en la síntesis del factor de crecimiento similar a la insulina tipo I (IGF-I), junto con una resistencia a la IGF-I y a otros factores de crecimiento atribuibles al hipercortisolismo, contribuye al retraso. Se ha descrito una reducción en la secreción de somatotropina (GH), basal y tras estímulos, antes y después del tratamiento quirúrgico de la enfermedad de Cushing. Por otra parte, los glucocorticoides actúan directamente en la placa epifisaria, ocasionando una reducción del crecimiento lineal21. Además, la virilización, con la consiguiente aceleración de la maduración ósea, puede resultar en una reducción del potencial de crecimiento incluso tras el tratamiento de la enfermedad.
Es probable que la edad de inicio y la duración de la hipercortisolemia antes de la curación sean factores que determinen la severidad del retraso y la talla final alcanzada.
Hay evidencia de que el efecto inhibitorio de la hipercortisolemia en la secreción de GH continúa durante al menos 1 o 2 años tras la curación de la enfermedad. La secreción de GH debe evaluarse precozmente (a diferencia de los adultos en los que se aconseja esperar 2 años tras el tratamiento quirúrgico de la enfermedad de Cushing)21 para, en caso de confirmarse el déficit, iniciar el tratamiento sustitutivo tan pronto como sea posible y así optimizar las posibilidades de crecimiento, además de normalizar la mineralización ósea22. Algunos autores recomiendan valorar la dinámica de secreción de GH a intervalos regulares23. No obstante, y a pesar del tratamiento sustitutivo, la mayoría de estos niños curados de la enfermedad no alcanzan la talla final adulta prevista.
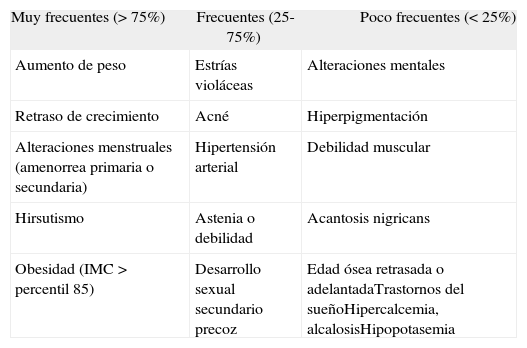
Otros síntomas por orden de frecuencia se muestran en la tabla 3.
Frecuencia con que ocurren los síntomas en el síndrome de Cushing en niños
| Muy frecuentes (> 75%) | Frecuentes (25-75%) | Poco frecuentes (< 25%) |
| Aumento de peso | Estrías violáceas | Alteraciones mentales |
| Retraso de crecimiento | Acné | Hiperpigmentación |
| Alteraciones menstruales (amenorrea primaria o secundaria) | Hipertensión arterial | Debilidad muscular |
| Hirsutismo | Astenia o debilidad | Acantosis nigricans |
| Obesidad (IMC>percentil 85) | Desarrollo sexual secundario precoz | Edad ósea retrasada o adelantadaTrastornos del sueñoHipercalcemia, alcalosisHipopotasemia |
Aunque los posibles cambios mentales no han sido sistemáticamente evaluados en niños, se ha descrito un rendimiento escolar satisfactorio, incluso con coeficiente intelectual por encima de lo normal, a diferencia de la disminución de capacidad de trabajo intelectual y físico observada en adultos. En este sentido, en contraste con lo observado en adultos, se ha objetivado un grado de inteligencia por encima de la media a pesar de tener una atrofia cerebral significativa. De hecho, la recuperación del eucortisolismo en estos niños se acompaña de un significativo deterioro de la función cognitiva, fundamentalmente verbal, a pesar de la completa recuperación de la atrofia cerebral24. En estudios de imagen secuenciales se objetiva un rápido incremento en el volumen cerebral, que alcanza la normalidad un año después del tratamiento quirúrgico. Estos cambios de volumen son significativamente mayores que los descritos en adultos tras la curación (la tasa de recuperación celular y neurogénesis puede diferir según la edad y el estado de desarrollo). Por el contrario, en adultos se describe un daño cognitivo y de memoria, así como alteraciones psicopatológicas significativas durante el hipercortisolismo, con una significativa recuperación de síntomas, pero sólo parcial de la atrofia cerebral después de conseguir el eucortisolismo. Los efectos psicológicos a largo plazo son desconocidos, pero no se ha descrito sintomatología depresiva en las series publicadas en niños24.
Contrariamente a lo que se cree respecto a la edad ósea de estos pacientes, ésta es congruente con la edad cronológica en el 81% de los casos, acelerada en un 8% y retrasada en el 11%, en clara relación con el estadio puberal del paciente.
Similarmente a lo que ocurre en adultos, el hipercortisolismo conlleva una disminución de la densidad ósea fundamentalmente en lugares donde predomina el hueso trabecular. En el caso de la enfermedad pediátrica, el mecanismo patogénico fundamental es la reducción de la actividad osteoblástica y de la síntesis de matriz ósea (por inhibición directa o indirecta de la función osteoblástica). Este aspecto es importante, ya que la masa ósea alcanzada durante la infancia es crucial en el máximo de masa que se alcanzará en la edad adulta. Aunque hay pocos datos respecto a la evolución de la densidad ósea tras el tratamiento, la recuperación de ésta parece depender de una pronta instauración del tratamiento sustitutivo para los déficit hipofisarios concomitantes, particularmente del déficit de GH25. No obstante, estos pacientes no llegan a alcanzar un máximo normal de masa ósea, y están expuestos a un aumento de riesgo de osteoporosis y, por lo tanto, de fracturas en la edad adulta26.
DiagnósticoPresenta mayores dificultades en estas edades. Las pruebas diagnósticas están basadas en estudios realizados en adultos y, luego, extrapolados a la edad pediátrica.
El protocolo diagnóstico más aceptado parte de una clínica indicativa (es mandatario realizar diagnóstico diferencial con la obesidad simple), con la dificultad añadida de que con frecuencia faltan datos de la velocidad de crecimiento.
La determinación de cortisol libre urinario en el cribado y la prueba con dosis bajas de dexametasona plantea la dificultad de la recogida adecuada, especialmente en los niños más pequeños. La medición de cortisol urinario debe ser corregida por el área de superficie corporal; se consideran valores normales<70μg/m2/día (198,7nmol/m2/día). No obstante, la determinación de concentraciones elevadas de cortisol en una muestra incompleta de orina, corregida por superficie corporal y creatinina, tiene un alto grado de sensibilidad (90-100%)26.
En la prueba de cribado de supresión con 1mg, después de evaluar varias dosis de dexametasona en niños normales, se ha indicado que la dosis adecuada podría ser 0,3mg/m2. Por esta discrepancia en la dosis óptima de dexametasona y la dificultad de la recogida adecuada de orina de 24h, hay autores que afirman que el test más eficaz de cribado es la medición del cortisol plasmático a las 24.00, a pesar de la dificultad de su realización ambulatoria. Una alternativa útil como cribado en niños es el cortisol salival nocturno (entre las 23.30 y las 24.00), que permite distinguir entre niños con hipercortisolismo y niños sanos, con una sensibilidad equivalente a la de la determinación del cortisol libre urinario (sensibilidad del 92-100% y especificidad del 95%), considerando valor normal≤7,5nmol/l. Sin embargo, las concentraciones de cortisol salival determinadas por la mañana se solapan entre niños afectados, no afectados y niños obesos, por lo que no es útil su medición a esa hora, como tampoco el estudio del ritmo circadiano con esta técnica (sensibilidad del 83%).
La combinación de la determinación nocturna de cortisol salival con la respuesta tras 1mg nocturno de dexametasona tiene altas sensibilidad (100%) y especificidad (93%) (valor de corte, 2,4nmol/l)27,28. En la supresión con dosis bajas de dexametasona la dosis aceptada es 20 μxg/kg/día (tasa de falsos negativos del 14%).
En lo referente al estudio radiológico en las series de pacientes publicadas, la resonancia magnética (RM) con gadolinio tiene una sensibilidad del 50 al 72%; presenta especiales problemas en niños pequeños porque precisan sedación, y en adolescentes porque con frecuencia llevan ortodoncia29.
Hay controversia sobre la conveniencia de realizar cateterismo de senos petrosos en estos pacientes. Según algunos autores, no suele ser necesario para confirmar el diagnóstico por la baja incidencia de incidentalomas hipofisarios a esta edad (menos del 6%), y en niños pequeños se debe considerar el riesgo añadido de la sedación o anestesia. Su indicación se reservaría únicamente para los casos en que la fuente de secreción de ACTH sea desconocida después de las pruebas habituales, y únicamente se debe realizar en centros experimentados (radiólogos intervencionistas con experiencia en pediatría) para evitar complicaciones y errores de interpretación30. Sin embargo, otros autores31 defienden que la exactitud de esta técnica en la localización preoperatoria del adenoma aconseja su realización sistemática, además de la radiología. Los criterios diagnósticos son los mismos que en adultos. El hallazgo de la lateralización concordante con los hallazgos quirúrgicos se produce en el 78% de los casos según los metaanálisis publicados32.
TratamientoAl igual que en adultos, en la enfermedad de Cushing el tratamiento de elección es la cirugía transesfenoidal, con una tasa de curación del 60 al 90%; se insiste en la necesidad de que este tipo de cirugía sea realizada por un neurocirujano experto30. La incidencia de rinorrea y meningitis es del 7-10%. Las dificultades técnicas en pacientes de esta edad se derivan del pequeño tamaño de la fosa hipofisaria y la ausencia de aireación del seno esfenoidal en algunos casos.
Si se confirma la persistencia de la enfermedad tras la cirugía, hay tres posibilidades de tratamiento: repetir la cirugía transesfenoidal (con riesgo elevado de déficit hipofisario permanente y con baja tasa de éxito), radioterapia hipofisaria y adrenalectomía unilateral o bilateral (incidencia de desarrollo de síndrome de Nelson del 25-67%). De ellas, la radioterapia hipofisaria ha quedado establecida como segunda opción de tratamiento tras la primera cirugía. Ha demostrado ser efectiva en niños, con unas tasas de curación del 80-88% (más elevada que en adultos). La dosis total es de 45Gy en 25 fracciones durante 35 días; el déficit de GH es una consecuencia inevitable. La respuesta es relativamente rápida, en 8 o 12 meses (en adultos, 1,5-4 años), con la necesidad de tratamiento médico durante ese periodo. La recuperación de la función hipófiso-adrenal ocurre en una media de 1,16 años31 tras la radioterapia. El déficit de GH tras tratamiento oscila entre el 85 y el 100% de los casos; se han descrito casos de recuperación espontánea de la secreción de esta hormona. Los déficit de otras hormonas son raros, pero los pacientes deben ser seguidos durante toda la infancia, la adolescencia y la juventud por la posibilidad de aparición tardía de un déficit hormonal hipofisario.
En la actualidad, no hay datos de la tasa de éxito de la radiocirugía en niños20.
Los criterios bioquímicos de curación no difieren de los establecidos en adultos.
El seguimiento tras la curación mediante cirugía consiste en valorar el crecimiento, la presión arterial, que debe normalizarse en los pacientes que desarrollaron hipertensión arterial, y la función tiroidea, ya que se describe hipotiroidismo secundario en el 17% de los pacientes intervenidos, en la mayor parte de los casos, transitorio20. Por la posibilidad de recidiva el seguimiento debe continuarse toda la vida.
SÍNDROME DE CUSHING EN EL EMBARAZOEl embarazo normal es un estado de relativo hipercortisolismo. Se produce un incremento progresivo del cortisol plasmático (total y libre) a partir de la semana 11 de gestación y hasta el parto, hasta 2 o 3 veces respecto al valor encontrado en controles no gestantes, de la proteína transportadora del cortisol (CBG) y del cortisol libre urinario (CLU) (el 180% comparado con valores en no gestantes)33. Esto parece deberse, por una parte, a un aumento de la función adrenal de la madre y de la unidad feto-placentaria, además de un aumento de producción de CBG por los estrógenos. También se ha indicado la influencia de un efecto antiglucocorticoideo periférico, producido por la progesterona, así como una alteración en el ajuste en el feedback negativo sobre la secreción de ACTH. La elevación progresiva de la ACTH objetivada indica una contribución de la secreción de CRH y de ACTH por la placenta.
La asociación de síndrome de Cushing y embarazo es muy poco frecuente. Su rareza se debe a que el hipercortisolismo habitualmente cursa con amenorrea, oligomenorrea e infertilidad36 por inhibición de la secreción de gonadotropinas. Se han descrito 136 gestaciones en 122 mujeres, con una edad gestacional al diagnóstico de 18,4±1 semanas34.
El síndrome de Cushing durante el embarazo conlleva morbilidad materna en el 70% de los casos. Las complicaciones más frecuentes son la hipertensión arterial (el 90% de los casos) y la diabetes mellitus (60%). Menos frecuentes son la osteoporosis, las fracturas, enfermedad psiquiátrica e insuficiencia cardíaca. No obstante, la muerte materna es rara; hay publicados 2 casos33,35,36.
En cuanto al feto, aunque la degradación placentaria del cortisol por la 11-beta-hidroxiesteroide deshidrogenasa 2 parece protegerlo del exceso de glucocorticoides, hay un aumento del riesgo de aborto espontáneo (20%), muerte perinatal, parto prematuro (en el 43% de los casos) y retraso de crecimiento intrauterino, con una mortalidad total fetal del 21%33,35.
La insuficiencia adrenal en el feto ocurre raramente. No obstante, tras el nacimiento, el neonato debe ser evaluado; se recomienda cobertura profiláctica con glucocorticoides. No se han descrito signos de exceso de glucocorticoides en el recién nacido.
EtiologíaDifiere respecto a la de la mujer no embarazada, ya que en el 40-50% de los casos es por causa adrenal (en contraste con el 15% de las mujeres no gestantes). El adenoma único es la causa más frecuente, mientras que el carcinoma es el 10% de los casos.
La enfermedad de Cushing es el 33% de los casos. Se han descrito 4 casos de secreción ectópica de ACTH por feocromocitoma y 8 casos por hiperplasia nodular pigmentada.
Se han descrito varios casos de síndrome de Cushing desarrollado sólo durante el embarazo, con remisión completa tras el parto; probablemente se trataba de hiperplasia nodular por expresión ectópica de receptores de LH en el córtex adrenal.
DiagnósticoDiagnosticar síndrome de Cushing durante el embarazo puede resultar difícil. Clínicamente ambas situaciones tienen en común un aumento de peso de distribución central, astenia, edemas, alteraciones emocionales (por ello es frecuente que el diagnóstico se retrase hasta las semanas 12–26 de la gestación). En ambas situaciones puede haber alteración del metabolismo de la glucosa e hipertensión arterial.
Desde el punto de vista bioquímico, también hay dificultades diagnósticas dado que no hay datos de cómo modificar los criterios bioquímicos diagnósticos durante el embarazo, y los tests empleados para diagnóstico diferencial no han sido evaluados sistemáticamente en esta situación.
Durante el primer trimestre la excreción de cortisol es similar a la de la mujer no embarazada. En el segundo y el tercer trimestre los valores se triplican, y se solapan con los de una mujer afecta no gestante. Por ello, parece establecerse un valor 3 veces por encima del límite superior de la normalidad como indicativo de síndrome de Cushing (hay una media de 8 veces de incremento, con unos límites de 2–22 veces), aunque hay controversia sobre cuál sería el límite superior normal en la gestación. Lo mismo ocurre con el valor límite de cortisol plasmático y salival de última hora de la tarde-noche.
Respecto al cortisol plasmático por la mañana, no es diferente del de la mujer embarazada normal, con una media de 37μg/dl. El ritmo circadiano de cortisol se mantiene durante la gestación, por lo que una ausencia de este ritmo es altamente indicativa de hipersecreción patológica.
La supresión con 1mg de dexametasona está disminuida durante el embarazo, lo que da un elevado porcentaje de falsos positivos, no por alteración de la biodisponibilidad de la dexametasona, sino por reajuste del feedback hipofisario, incremento de la CBG y la resistencia tisular a los glucocorticoides por la progesterona36,37. Esta falta de supresión persiste hasta 4–5 semanas tras el parto.
Por ello, se ha recomendado la combinación de una elevación de 3 veces el límite superior de la normalidad en el cortisol libre urinario con elevación en el cortisol salival nocturno como estrategia de cribado.
En el diagnóstico diferencial entre hipercortisolismo dependiente e independiente de ACTH la valoración de la ACTH plantea la dificultad de que en el 50% de los casos de causa adrenal la ACTH no está suprimida, en parte por una continuada estimulación del eje hipofisario-adrenal por el CRH placentario, y en parte por la secreción placentaria de ACTH. Si la ACTH está suprimida o si la concentración de ACTH es mayor de 15pg/ml, no son necesarias más pruebas bioquímicas.
Cuando la concentración de ACTH no es concluyente, la prueba de supresión con dosis altas de dexametasona, aunque no ha sido formalmente validada en el embarazo, puede ayudar a discriminar entre causa adrenal o hipofisaria, con la limitación de no diferenciar todos los casos de enfermedad hipofisaria. Su utilidad para el diagnóstico diferencial de secreción de ACTH ectópica en el embarazo es desconocida por los pocos casos comunicados.
La prueba de CRH quedaría limitada a casos de duda, dado que el CRH ovino está incluido en la categoría C de la Food and Drug Administration (FDA). La respuesta de ACTH parece estar disminuida en el tercer trimestre por lo que su utilidad también estaría reducida, y no hay suficientes estudios sistemáticos.
Se han publicado varios casos de cateterismo de senos petrosos inferiores en mujeres gestantes, en que la cateterización se ha realizado por vía yugular para minimizar la radiación fetal, sin incidencias35. Quedaría limitado a los casos no diagnosticados, tras agotar los métodos no invasivos y en centros experimentados.
En lo referente a la radiología, en el caso del síndrome de Cushing hipofisario, la resonancia es la única prueba a realizar. No obstante, está contraindicada en el primer trimestre por su posible (pero no probado) potencial teratogénico; se considera segura después de la semana 32. En el segundo trimestre y hasta esta fecha, debe valorarse la relación riesgo-beneficio en cada paciente. Hay que tener en cuenta que dado que la RM detectará un tumor incidental (≤ 6mm) en el 10% de los individuos sanos, y la hipófisis duplica su tamaño en el tercer trimestre de la gestación, podría haber mayor proporción de incidentalomas. El contraste paramagnético gadolinio es categoría C de la FDA, por lo que se aconseja no utilizarlo, reduciéndose la sensibilidad de esta prueba al 38%.
Respecto al Cushing de origen adrenal, la ecografía tiene una sensibilidad del 70-97% en la detección de lesiones adrenales; su utilización en el embarazo no presenta ninguna limitación. La resonancia magnética sería la segunda opción, que se prefiere sobre la tomografía computarizada, por la radiación de esta última.
En resumen, como algoritmo diagnóstico se ha propuesto la combinación de cortisol libre urinario y cortisol salival nocturno para cribado. En pacientes con confirmación del hipercortisolismo, la determinación de concentraciones de ACTH bajas indicaría causa adrenal y hay que realizar pruebas de imagen. Si la ACTH está en el límite, una combinación de prueba de supresión con 8mg y prueba de CRH distinguiría las diferentes formas de dependencia de ACTH.
El algoritmo diagnóstico del síndrome de Cushing durante el embarazo se esquematiza en la figura 1.
Tratamiento
Se recomienda tratamiento quirúrgico, excepto cuando el diagnóstico se realiza al final del primer trimestre. La cirugía debe realizarse durante el segundo trimestre de gestación.
Aunque todo procedimiento quirúrgico supone riesgo, el riesgo derivado de no intervenir es considerablemente más alto. Incluso en casos aparentemente en remisión tras tratamiento, la incidencia de eclampsia y parto prematuro es más elevada.
El planteamiento quirúrgico no es diferente durante la gestación. En el caso del síndrome de Cushing adrenal es preferible la vía laparoscópica si no hay contraindicación.
La adrenalectomía bilateral sería la opción para pacientes que requieren una inmediata curación, en casos de tumor primario oculto, inoperable o metastásico, al igual que en pacientes no embarazadas.
En el caso del Cushing de origen hipofisario, la cirugía transesfenoidal es el tratamiento de elección; no se han descrito diferencias respecto a pacientes no gestantes.
El tratamiento médico estaría indicado en casos diagnosticados al final del embarazo o durante la preparación para la cirugía. En el tratamiento médico la metopirona es bien tolerada, y aunque los principales efectos secundarios son hipertensión y preeclampsia (los cuales limitan su utilización), es el fármaco de elección dentro de esta forma de tratamiento. El ketoconazol está incluido en la categoría C de la FDA, por lo que su empleo debe limitarse a casos de intolerancia a la metopirona (en animales se ha demostrado efecto antiandrogénico y teratógeno en el feto, aunque no hay casos descritos en humanos). La ciproheptadina, aunque segura, no es efectiva y no debe indicarse. Con mitotano se ha demostrado teratogenicidad y con aminoglutetimida se ha descrito virilización fetal.
