




El aumento de la morbimortalidad cardiovascular en estados de resistencia insulínica (RI), como la obesidad, el síndrome metabólico, y la diabetes tipo 2, representa un problema mayor para la salud pública. La dislipemia de la RI comprende la hipertrigliceridemia, una disminución de colesterol de las lipoproteínas de alta densidad, aumento de las lipoproteínas de alta densidad pequeñas y densas e hiperlipemia posprandial, cumpliendo un papel directo e indirecto en la arterosclerosis. Esta dislipemia es debida a la acumulación de lipopartículas ricas en triglicéridos de origen intestinal y hepático. El intestino ha sido considerado un órgano pasivo, pero la evidencia actual confirma al intestino como un órgano activo sometido a la regulación de: ácidos grasos libres, insulina, incretinas e inflamación. Dos conceptos han surgido, el de la RI intestinal y el de la sobreproducción de quilomicrones en los estados de hiperinsulinismo/RI. Una comprensión de la RI intestinal puede convertit al enterocito en una diana terapéutica.
The increase in cardiovascular morbidity and mortality associated to insulin resistance (IR) states (obesity, metabolic syndrome, type 2 diabetes) represents a major public health problem. In IR, dyslipidemia typically include hypertriglyceridemia, low high density lipoprotein cholesterol, increased small and dense low density lipoprotein particles, and post-prandial hyperlipidemia, which play a direct or indirect role in the mecanisms of atherosclerosis. Dyslipidemia is mainly due to accumulation of circulating triglyceride-rich lipoproteins from the liver and bowel. The bowel has traditionally been seen as a passive organ, but current evidence confirms that it is an active organ subject to regulation by free fatty acids, insulin, incretins, and inflammation. Two new concepts have emerged: intestinal IR and overproduction of chylomicrons in hyperinsulinemic/IR states. A better understanding of intestinal IR may make the enterocyte a therapeutic target.
El aumento alarmante de la prevalencia de la resistencia insulínica (RI) en patologías como la diabetes tipo 2, la hiperlipidemia familiar combinada (HPFC) y el síndrome metabólico, en asociación con la actual epidemia de la obesidad, representan un problema vital para la salud pública1. La aterosclerosis y sus complicaciones coronarias, cerebrales y vasculares periféricas constituyen la primera causa de morbimortalidad en estados de RI, con un riesgo relativo de 2 a 3 en los hombres y de 4 a 5 en las mujeres que padecen diabetes mellitus tipo 22.
Los trastornos lipídicos juegan un rol preponderante en la enfermedad cardiovascular asociada a los estados de RI. El principal factor de riesgo lipídico es el colesterol de las lipoproteínas de baja densidad (C-LDL). Esta observación ha sido confirmada en estudios epidemiológicos y estudios de intervención perfectamente concordantes3. La dislipemia de la RI está caracterizada por el siguiente cuarteto: hipertrigliceridemia en ayunas, baja concentración de colesterol de las lipoproteínas de alta densidad (C-HDL), aumento del número de partículas de LDL pequeñas y densas e hiperlipemia posprandial4. Cada uno de estos 4 parámetros constituye marcadores de riesgo cardiovascular independientes5–9. Existen numerosos argumentos que sostienen la hipótesis de que el riesgo cardiovascular no desaparece totalmente con bajos niveles de C-LDL, en particular en patologías como la diabetes, definiendo lo que actualmente se llama riesgo cardiovascular residual10.
Algunos autores atribuyen parte del proceso aterogénico a una mayor producción de triglicéridos (TG) provenientes del intestino en el periodo posprandial9,11. En efecto, la hiperlipemia posprandial ha sido identificada como factor de riesgo vascular en estudios de cohorte y de caso-control12,13. Estudios recientes indican que los pacientes diabéticos tipo 2 presentan un aumento de los niveles plasmáticos de lipoproteínas ricas en triglicéridos (LRT) de origen intestinal en ayunas y en estado posprandial14,15; una asociación significativa ha sido encontrada entre la lipemia posprandial y la aterosclerosis coronaria16. Indefectiblemente, los remanentes de LRT intestinales son capaces de entrar al espacio subendotelial de la pared vascular y participar en el proceso de aterosclerosis17. El objetivo del presente trabajo es el de resumir los recientes descubrimientos referentes al nuevo rol del enterocito en relación a la fisiopatología de la dislipemia del estado de RI.
Papel de lipoproteínas ricas en triglicéridos en la aterogénesisVisión clásicaEn el hombre, la apolipoproteína (apo) B-48 es sintetizada a nivel del intestino delgado a partir del ARNm de la apoB-100 por un mecanismo postranscriptional llamado «mRNA editing», necesario para la síntesis, el ensamblaje y la secreción del quilomicrón (QM)18–20. Existe solo una molécula de apoB-48 por QM21; así es posible, en el hombre, distinguir las LRT de origen hepático portadoras de la apoB-100 (LRT-apoB-100) de las LRT de origen intestinal portadoras de apoB-48 (LRT-apoB-48). El contenido de apoB-48 es determinante del número de partículas de LRT intestinales y de sus remanentes, teniendo estas últimas mayor poder aterogénico. En cambio, el contenido en TG es un determinante del tamaño de la partícula; un mayor tamaño disminuye su potencial aterogénico al no poder atravesar el espacio subendotelial22.
La dislipemia de sujetos con RI es en gran parte debida a la acumulación plasmática de LRT hepáticas e intestinales. Esta acumulación es a la vez secundaria a la hiperproducción hepática de las lipoproteínas de muy baja densidad (VLDL) de tipo 1 (grandes y ricas en TG)23, a un defecto de depuración de las LRT que pueden ser atribuidos a la disminución de la actividad de la lipoproteína lipasa (LPL)24,25, a una anomalía de la composición apoproteica de las lipoproteínas26, al aumento del pool circulante de LRT plasmáticas resultado de la competencia entre las LRT hepáticas e intestinales que utilizan la misma vía de depuración saturable de la LPL27, y a un defecto de la captación hepática28.
El aumento del tiempo de permanencia en plasma de las LRT facilita la transferencia de TG de estas partículas a LDL y HDL, por acción de la proteína transportadora de colesterol esterificado (CETP); las LDL y HDL enriquecidas en TG son sustratos privilegiados de la lipasa hepática (LH), cuya acción es responsable de la formación de LDL pequeñas y densas, y del aumento de catabolismo de las HDL23. Las LRT actúan indirectamente en los fenómenos de aterosclerosis favoreciendo la aparición de 2 anomalías lipídicas aterogénicas: el aumento del número de LDL pequeñas y densas, y la baja de concentración de C-HDL (fig. 1).
 Enriquecimiento de lipoproteínas de baja densidad (LDL) y lipoproteínas de alta densidad (HDL) en triglicéridos (TG) por medio de la proteína transportadora de colesterol esterificado (CETP); 2) hidrólisis de TG por la lipasa hepática (LH); 3) aumento de LDL pequeño y denso (LDL-pd) y disminución de HDL por hipercatabolismo; 4) enriquecimiento de leucocitos/monocitos en ácidos grasos libres; 5) activación del endotelio; 6) fijación de remanentes de quilomicrón (r-QM) a los proteoglicanos; 7) acumulación de r-QM; 8) fijación de r-QM en los receptores de apoB-48 de macrófagos; 9) transformación de macrófagos en células espumosas; 10) migración y proliferación de células musculares lisas de la media. Col-E: colesterol esterificado.")
Implicación de lipopartículas ricas en triglicéridos intestinales en la aterosclerosis.
1) Enriquecimiento de lipoproteínas de baja densidad (LDL) y lipoproteínas de alta densidad (HDL) en triglicéridos (TG) por medio de la proteína transportadora de colesterol esterificado (CETP); 2) hidrólisis de TG por la lipasa hepática (LH); 3) aumento de LDL pequeño y denso (LDL-pd) y disminución de HDL por hipercatabolismo; 4) enriquecimiento de leucocitos/monocitos en ácidos grasos libres; 5) activación del endotelio; 6) fijación de remanentes de quilomicrón (r-QM) a los proteoglicanos; 7) acumulación de r-QM; 8) fijación de r-QM en los receptores de apoB-48 de macrófagos; 9) transformación de macrófagos en células espumosas; 10) migración y proliferación de células musculares lisas de la media. Col-E: colesterol
esterificado.
Las LRT pueden actuar directamente en el proceso de aterosclerosis con el enriquecimiento en ácidos grasos libres circulantes (AGL), que son ligandos endógenos de receptores tipo Toll presente en los leucocitos29, conduciendo a la activación del factor de transcripción nuclear Kappa B (NFkB)29, generando de esta forma la síntesis de citoquinas inflamatorias y procoagulantes, manteniendo así un microambiente proinflamatorio procoagulante y proaterogénico en la pared arterial con sus consecuencias deletéreas11 (fig. 1).
Una acción más directa pasa por la retención de los remanentes de las LRT hepáticas e LRT intestinal en la pared arterial. En efecto, un sitio de unión de la apoB-48ha sido identificado en los proteoglicanos de la íntima arterial30. Además, se aisló y caracterizó en macrófagos y monocitos un receptor específico de la apoB-4831,32. Este receptor (apoB-48-R) difiere de los receptores de LDL apoB/E, LRP (LDL-receptor related protein) y receptores basureros (scavengers), en que capta únicamente las LRT-apoB-48 provenientes del intestino. Este receptor está presente a nivel de las células espumosas en las estrías lipídicas y en las placas de ateroma. Es probable que su capacidad de enlace y de internalización de remanentes de LRT intestinales participe en la adquisición del fenotipo «foam cell» de los macrófagos y en la constitución de las lesiones de aterosclerosis (fig. 1). Recientemente, la apoB-48ha sido aislada de placas aórticas de ateroma en el humano33. La capacidad de retención de los remanentes de LRT-apoB-100 es 10 veces superior, teniendo en cuenta su concentración plasmática en relación a los remanentes de LRT-apoB-48, pero la concentración en colesterol es 40 veces superior en las LRT-apoB-48 con relación a las LRT-apoB-100. Esta condición otorga a las LRT-apoB-48 la posibilidad de liberar 4 veces más colesterol en la pared vascular que las LRT-apoB-10017.
Por todos estos datos, las LRT-apoB-48 de origen alimentario, QM y su remanente, parecen tener poder aterogénico. Datos obtenidos a partir de estudios en animales permiten corroborar este papel aterogénico. De hecho, existen modelos de ratones transgénicos (ApoCIII+/+ y ApoE-/-) que se caracterizan por una elevación importante de las partículas lipoprotéicas portadoras de apoB-48 (aunque en el ratón la apoB-48 no es específica del intestino). Estos animales sufren una aterogénesis acelerada sobre todo si el contenido lipídico proveniente de su alimentación está aumentado34,35.
De lo que precede, podemos concluir que, aparte de la LDL (y en particular la subfracción pequeña y densa), existen otras lipopartículas aterogénicas: QM y su remanente de origen alimentario, teniendo la apoB-48 no solo un papel como marcador de QM sino también un papel activo en la formación de la placa de arteriosclerosis. La visión clásica de la acumulación sanguínea de LRT-intestinales es secundaria a la disminución del aclaramiento de QM en los estados de RI.
Exploración de la lipemia posprandialEl metabolismo de las LRT intestinales se puede analizar por medio de 2 métodos: el primero es el área bajo la curva (ABC) de marcadores del QM como la vitamina A y/o la apoB-48 posterior a una sobrecarga oral en lípidos36(SOL), y el segundo el método cinético de la apoB-48 utilizando la perfusión de isótopos estables en condiciones de equilibrio39(plateau).
En el primer método se utiliza un metabolito de la vitamina A, el retinol palmitato (RP) puesto que es incorporado al QM, y luego es recaptado por el hígado sin ser secretado ulteriormente37. La apoB-48 es secretada únicamente en el intestino y no es intercambiable con otras fracciones lipídicas, lo cual la constituye en un útil marcador de QM38. La diferencia consiste en que el RP es intercambiable en un 18-25% con LDL y se incorpora solo en el QM maduro, no se integra en el QM recién formado; estas 2 diferencias no existen utilizando la apoB-48 como marcador específico intestinal36.
El segundo método, llamado cinético, constituye el gold standard para estudiar el metabolismo de QM, y consiste en administrar productos nutricionales cada 30 o 60 minutos adaptados a la necesidades calóricas cotidianas de cada paciente, con el propósito de estimular la síntesis de apoB-48, obteniendo una concentración plasmática estable de apoB-4839,40(plateau). Paralelamente a la alimentación, se acopla la perfusión de un isótopo estable, como es el caso del aminoácido leucina marcado con deuterio (2H3-leucina) o con carbono 13 (1-13C-leucina), a fin de incorporarse a la apoB-48 neo-sintetizada. El objetivo es obtener un cociente entre la apoB-48 que incorporó el isótopo estable y la apoB-48 que no incorporó el isótopo estable. Este cociente se expresa en porcentaje, obteniendo así una curva de enriquecimiento isotópico; estos valores serán incorporados en un modelo matemático multicompartimental, a fin de calcular la tasa de producción (TP) y la tasa de catabolismo (TC) de la apoB-4841. Resumiendo, la metodología empleada en la SOL se obtiene el ABC de la apoB-48 es la técnica más simple pero la más imprecisa; en cambio, en la técnica de cinética se obtiene la TP y la TC de la apoB-48, es más exacta aunque más compleja a realizar.
Regulación de la síntesis de lipoproteínas ricas en triglicéridos intestinalesModulación de la alimentaciónLa producción de las LRT intestinales ha sido considerada durante mucho tiempo regulada por el aporte alimentario y, más específicamente, por la cantidad de lípidos ingeridos. Numerosos estudios se han interesado en la relación entre el aporte alimentario y la lipemia posprandial sin hacer diferencia entre las anomalías de producción y/o de depuración del QM y su remanente. Las raciones lipídicas de 20 a 50g por comida aumentan de manera dosis dependiente la lipemia posprandial, y con raciones superiores a 80g por comida el aumento es constante42. El agregado de sacarosa y/o fructosa a una comida rica en grasas o de glúcidos con fuerte índice glucémico aumenta la lipemia posprandial42.
Los otros factores alimentarios que pueden jugar un rol leve a moderado en la reducción de la lipemia posprandial corresponden a la administración de ácidos grasos poliinsaturados n-3 (omega-3), n-6 (omega-6) o a alimentos ricos en fibras42.
El aumento de la lipemia posprandial también puede ser secundario a una comida pobre en colesterol o rica en proteínas43. El aporte alimentario contribuye a un aumento marcado del contenido de TG más que del contenido de apoB-48 en la partícula de QM, con el respectivo aumento del tamaño de las partículas de LRT más que del número de las mismas22.
Hiperproducción de lipoproteínas ricas en triglicéridos intestinales en estados de resistencia insulínicaModelo animalPara estudiar el rol de la producción de las LRT intestinales en el desarrollo de la dislipemia metabólica, el modelo animal de Syrian golden hamster (fructose-fed hamster), convertido en resistente a la insulina gracias a una alimentación rica en fructosa, constituye un modelo de estudio útil44. Como en el hombre, la expresión tejido-específica de la apoB-100 únicamente en el hígado y de la apoB-48 únicamente en el intestino representa una ventaja en el modelo del hámster, permitiendo estudios in vivo de la producción de las LRT intestinales vs. las LRT hepáticas45,46. En este modelo, se ha demostrado un aumento de la producción de LRT 2-4 veces mayor que en el modelo de hámster sin RI. Esta secreción aguda de LRT intestinales ha sido confirmada ex vivo en cultivos de enterocitos con RI. De manera similar a la hiperproducción de LRT hepáticas47–49, las experiencias ex vivo e in vitro sobre el enterocito han mostrado que la alimentación crónica con fructosa está asociada a una mayor estabilidad de la apoB-48 intracelular, a una mayor lipogénesis de novo, a un aumento de la síntesis endógena de TG y de ésteres de colesterol, y a una mayor expresión de la proteína de transferencia microsomal (MTP), proteína clave en la formación de lipoproteínas intestinales44 (fig. 2). Una alimentación rica en fructosa de solo 2 días no produce ningún efecto sobre la producción de LRT intestinales, interpretando que el efecto de esta alimentación rica en fructosa es de tipo crónico44.

Mecanismos de híperproducción de lipopartículas ricas en triglicéridos intestinales en un contexto de hiperinsulinismo y resistencia insulinica.
AGL: ácidos grasos libres; AGω3: ácidos grasos omega-3; FAT/CD36: translocasa de ácidos grasos; GLP-1: péptido-1 similar al glucagón; GLP-2: péptido-2 similar al glucagón; MTP: proteína de transferencia microsomal; SREBP-1c: elemento de respuesta a esteroles 1c; TNFα: factor de necrosis tumoral alfa; TG: triglicéridos.
Esta hiperproducción de LRT intestinales ha sido confirmada, in vivo, ex vivo e in vitro, en otro modelo de RI, el high fat-fed Syrian Golden hamster50. Un tratamiento con rosiglitazona (un agonista del receptor activado por proliferadores peroxisómicos tipo alfa (PPAR-γ)) en estos 2 modelos animales de hámster con RI corrige la elevación de la concentración de MTP y reduce la sobreproducción intestinal de LRT, probablemente favoreciendo la sensibilidad a la insulina50,51. Los resultados de este modelo han sido muy similares al modelo con RI/diabetes tipo 2 (gerbille psammomys obesus) corroborando la asociación entre la RI y la sobreproducción intestinal de LRT. En este modelo, el aumento de la monoacilglicerol transferasa (MGAT), de la diacilglicerol transferasa (DGAT) y de la expresión de la proteína de unión de ácidos grasos L (L-FABP) ha favorecido la lipogénesis de novo, sin encontrar anomalías en la expresión de MTP52.
En relación a las vías de señalización de la insulina, las experiencias in vivo, ex vivo e in vitro, realizadas en el modelo de hámster con RI (fructose-fed hamster) han mostrado defectos a nivel enterocitario, implicando una disminución del sustrato del receptor de insulina-1 (IRS-1) e inhibición de la proteína quinasa B o AkT, e inversamente un aumento de la concentración de la subunidad p110 de la PI-3 kinasa, de la tirosina fosfatasa 1-B, y de la vía de proteínas quinasas activadas por mitógenos (MAPK/ERK). Estas anomalías están a favor de una RI intestinal, que provoca una hiperactivación de la vía MAP/ERK, conduciendo a un aumento de la expresión de la MTP, de la apoB, y de un factor de transcripción, el elemento de respuesta a esteroles 1c (SREBP-1c), que estimula la síntesis de proteínas que participan de la lipogénesis de novo53,54. El conjunto de estas anomalías conduce a un aumento de producción y secreción de LRT intestinales53,54 (fig. 2).
Modelo humanoLa sobreproducción de las LRT intestinales es reconocida como un nuevo componente del síndrome de RI. Este exceso de producción ha sido confirmado por primera vez en el hombre, en el cual la producción intestinal de LRT está aumentada en función del grado de hiperinsulinismo y de sensibilidad a la insulina41. A diferencia de los modelos animales, en el sujeto sano la rosiglitazona no mejora la producción de LRT intestinales, sino que la empeora a pesar de su efecto insulinosensibilizador, con una tendencia no significativa al aumento de producción y a la reducción de la depuración de las LRT intestinales55 (tabla 1). Estos resultados podrían explicar el aumento de aterosclerosis coronaria en pacientes tratados con rosiglitazona56. Estudios observacionales de pacientes obesos con RI han mostrado un aumento del nivel plasmático de apoB-48 en ayunas y en estado posprandial57,58. En los pacientes diabéticos tipo 2, la hiperproducción de LRT intestinales se encontró asociada a una disminución de su depuración14. La HPFC se caracteriza por mayor concentración de apoB-48 en ayunas y en estado posprandial59,60 (tabla 2). Es bien conocido que la acumulación de apoB-48 se produce por disminución de la actividad LPL secundaria al aumento de LRT-hepática60 asociado a su vez al aumento de la apoC-III (inhibidor fisiológico de la LPL). A su vez, la apoC-III estimula la producción de LRT participando de la etapa final del ensamblaje de LRT a nivel celular61. La presencia de esteatosis hepática en la diabetes tipo 2 y en la HPFC se asocia a un aumento de la producción de apoB-48 en el estado posprandial62,63. El estado de RI es el determinante principal de la hiperproducción de apoB-48, con implicación activa en la enfermedad coronaria de estos pacientes64.
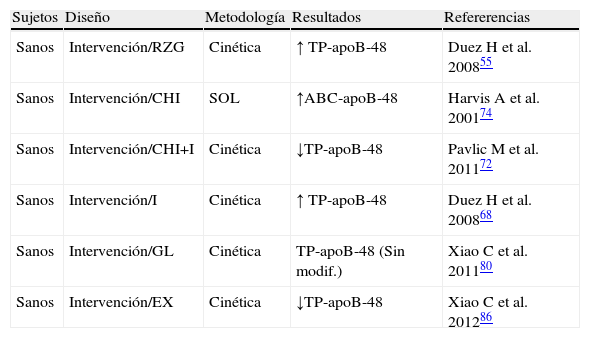
Exploración del metabolismo de lipoproteínas ricas en triglicéridos (LRT)-apoB-48 en estados de sensibilidad a la insulina conservada
| Sujetos | Diseño | Metodología | Resultados | Refererencias |
| Sanos | Intervención/RZG | Cinética | ↑ TP-apoB-48 | Duez H et al. 200855 |
| Sanos | Intervención/CHI | SOL | ↑ABC-apoB-48 | Harvis A et al. 200174 |
| Sanos | Intervención/CHI+I | Cinética | ↓TP-apoB-48 | Pavlic M et al. 201172 |
| Sanos | Intervención/I | Cinética | ↑ TP-apoB-48 | Duez H et al. 200868 |
| Sanos | Intervención/GL | Cinética | TP-apoB-48 (Sin modif.) | Xiao C et al. 201180 |
| Sanos | Intervención/EX | Cinética | ↓TP-apoB-48 | Xiao C et al. 201286 |
ABC-apoB-48: área bajo la curva de la apoproteína B48; CHI: clamp hyperinsulinémico; EX: exenatide; GL: glucagon; I: intralipidos; RZG: rosiglitazona; Sin modif.: sin modificación significativa; SOL: sobrecarga oral en lípidos; TP-apoB-48: tasa de producción de la apoproteína B48.
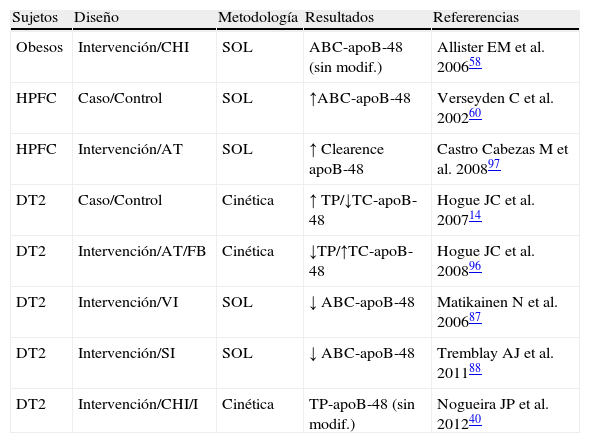
Exploración del metabolismo de lipoproteínas ricas en triglicéridos (LRT)-apoB-48 en estados de resistencia a la insulina
| Sujetos | Diseño | Metodología | Resultados | Refererencias |
| Obesos | Intervención/CHI | SOL | ABC-apoB-48 (sin modif.) | Allister EM et al. 200658 |
| HPFC | Caso/Control | SOL | ↑ABC-apoB-48 | Verseyden C et al. 200260 |
| HPFC | Intervención/AT | SOL | ↑ Clearence apoB-48 | Castro Cabezas M et al. 200897 |
| DT2 | Caso/Control | Cinética | ↑ TP/↓TC-apoB-48 | Hogue JC et al. 200714 |
| DT2 | Intervención/AT/FB | Cinética | ↓TP/↑TC-apoB-48 | Hogue JC et al. 200896 |
| DT2 | Intervención/VI | SOL | ↓ ABC-apoB-48 | Matikainen N et al. 200687 |
| DT2 | Intervención/SI | SOL | ↓ ABC-apoB-48 | Tremblay AJ et al. 201188 |
| DT2 | Intervención/CHI/I | Cinética | TP-apoB-48 (sin modif.) | Nogueira JP et al. 201240 |
ABC-apoB-48: área bajo la curva de la apoproteína B48; AT: atorvastatina; CHI: clamp hyperinsulinémico; FB: fenofibrato; HPFC: hiperlipidemia familiar combinada; I: intralípidos; Sin modif.: sin modificación significativa; SI: sitagliptina; SOL: sobrecarga oral en lípidos; TP-apoB-48: tasa de producción de la apoproteína B48; VI: vildagliptina.
La participación del enterocito en el metabolismo lipídico y en la producción de LRT intestinales, reconocidas como un nuevo componente de la RI y como actores destacados, en la etiopatogenia de la aterosclerosis, ha justificado la búsqueda de factores que modulen esta producción.
Transportadores intestinales de lípidosDurante mucho tiempo, la difusión pasiva ha sido considerada la vía de entrada principal de nutrientes en la célula intestinal. La puesta en evidencia de transportadores proteicos específicos sobre la membrana microvellositaria ha cambiado este antiguo concepto. Entre los principales, se pueden destacar el fatty acid translocase (FAT/CD36), implicado en la absorción de colesterol y ácidos grasos de cadena larga, la Niemann Pick C1 like 1 (NPC1L1) implicada en la absorción de colesterol y fitoesterol, ATP-Binding-Cassette transporter A1 (ABCA1), implicado en el flujo de colesterol, y el ATP-Binding-Cassette transporter G5/G8 (ABCG5/G8), implicado en el flujo de esteroles. El contenido enterocitario de lípidos es un regulador de la producción de los quilomicrones. Una ilustración de esta condición es el modelo de rata deficitaria en transportador FAT/CD36 que presentan una reducción de la secreción linfática de TG65. En los modelos de ratas diabéticas (inducido por la estreptozotocina) en comparación con ratas controles, el ARNm intestinal de la NPC1L1 y de la MTP aumenta, mientras que el ABCG5 y el ABCG8 disminuyen, correlacionando positivamente con los niveles de TG y colesterol dentro de los QM y con la concentración de apoB-484.
En un estudio llevado a cabo en pacientes diabéticos tipo 2 y en controles, se realizó biopsia duodenal y se observó que el ARNm de la NPC1L1, de la MTP y el ABCG5/8 se correlacionaban positivamente con los niveles de TG, demostrando una asociación entre la expresión de genes enterocitarios y las anomalías lipídicas en la diabetes tipo 266.
Ácidos grasos libresLos AGL circulantes están aumentados en la diabetes tipo 2, la obesidad y la HPFC63. Estos AGL se acumulan en tejidos extra-adipocitarios ejerciendo diferentes efectos deletéreos bajo la denominación común de lipocitotoxicidad. El flujo de AGL a nivel hepático estimula la producción y secreción de LRT67, principalmente en la RI, ya que la oxidación hepática de ácidos grasos está reducida. La elevación plasmática aguda de AGL estimula la producción de LRT intestinales y hepáticas sin modificación de la depuración en el sujeto sano68 (tabla 1). Este efecto estimulante de los AGL sobre la producción de LRT ha sido demostrado previamente en el modelo de Syrian golden hamster insulinosensible sin efecto aditivo en el modelo de hámster resistente a la insulina, excepto cuando ha sido tratado con insulinosensibilizadores como la rosiglitazona51. El mecanismo que explica este fenómeno es un bloqueo de la vía de señalización insulínica, una estabilización de la apoB-48, una incorporación directa de AGL a las lipoproteínas, una estimulación de la movilización y la incorporación de TG de reserva, y un aumento de la síntesis de novo de TG51(fig. 2).
Un estudio efectuado in vivo, ex vivo e in vitro en la rata ha demostrado que el efecto de los AGL a nivel hepático es de tipo bimodal; los AGL a pequeñas concentraciones y durante corto tiempo estimulan la secreción de LRT hepáticas, mientras que los AGL a grandes concentraciones y durante un tiempo prologando disminuyen la producción de LRT hepáticas69. Corroboramos este efecto supresivo de los AGL sobre la producción de LRT hepáticas en los pacientes diabéticos tipo 2 bajo clamp euglucémico hiperinsulinémico posterior a la perfusión de intralipid y heparina40. Este efecto bimodal de los AGL estuvo ausente en relación a la apoB-48 en el modelo animal y en la población de pacientes diabéticos tipo 240,69.
InsulinaContrariamente al efecto estimulante sobre la producción de LRT hepáticas e intestinales del hiperinsulinismo asociado a la RI4,23, está generalmente aceptado que la elevación aguda de la concentración de insulina inhibe la secreción de LRT hepáticas e intestinales. Esto ha sido demostrado en el hombre, in vivo70–72 e in vitro73. En el hombre sano, Malmström et al. han demostrado que el efecto inhibidor de la insulina sobre la producción de LRT hepáticas es independiente del efecto supresor de la insulina sobre los AGL70; en cambio, Pavlic et al. han mostrado que el efecto inhibidor de la insulina sobre la producción de LRT intestinales es parcialmente dependiente del efecto de la insulina sobre los AGL72 (tabla 1).
En el hombre sano, un hiperinsulinismo inducido por alimentos con un importante índice glucémico o por un clamp euglucémico hiperinsulinémico, retrasa la aparición de LRT intestinales en el plasma después de una comida rica en grasas74 (tabla 1). Un estudio realizado in vitro ha mostrado que la adición aguda de insulina en medio de cultivo de células fetales humanas de intestino reduce la secreción de quilomicrones75. Los pacientes diabéticos tipo 2, obesos y con HPFC, que presentan un hiperinsulinismo crónico secundario a la RI no responden al efecto inhibitorio agudo de la insulina sobre la producción del LRT hepáticas, probablemente secundarios a la presencia de esteatosis hepática71,76,77. Comprobaciones idénticas han sido realizadas en hepatocitos de ratas con RI78. La acción supresiva de la insulina sobre la producción de QM está ausente en los pacientes obesos con RI sometidos con un clamp hiperinsulinémico58. En un trabajo previo, acabamos de demostrar que los pacientes diabéticos tipo 2 bajo clamp euglucémico hiperinsulinémico son resistentes al efecto inhibitorio agudo de la insulina sobre la producción del LRT intestinales40 (tabla 2).
GlucagónEsta hormona pancreática puede participar en el metabolismo oxidativo de los AGL a nivel hepático79. En ratas, el glucagón disminuye la síntesis y secreción de TG, y activa la β-oxidación por medio de la activación de PPAR-α79. En el hombre sano, la administración aguda de glucagón disminuye la secreción y catabolismo de VLDL tipo 1, sin modificación en su concentración plasmática; en cambio, la secreción y catabolismo del QM no se modificó significativamente80 (tabla 1). La ausencia de efecto del glucagón sobre el enterocito pude deberse a la disminución de receptores al glucagón en el intestino en comparación con el hígado81.
IncretinasEl péptido-1 similar al glucagón (GLP-1) y el péptido-2 similar al glucagón (GLP-2) son secretados en respuesta al consumo oral de alimentos, sobre todo los ricos en grasas e hidratos de carbono. La secreción del GLP-1, en el caso del hombre, puede ser estimulada por el ácido oléico82, sugiriendo una asociación entre el metabolismo lipídico y el GLP-1. La idea de que las incretinas modulan el metabolismo de los quilomicrones surge en el año 2005, cuando se observó que la administración de GLP-1 recombinante en ratas sanas disminuyó la secreción intestinal de TG83. La administración de un inhibidor de dipeptidil peptidasa iv (DPP-IV), como la sitagliptina, disminuye la secreción de TG en el modelo de syrian golden hamster. Este modelo permite explicar el nexo entre la señalización insulínica y el efecto de GLP-184.
La administración de GLP-1 en el hombre sano disminuyó la lipemia posprandial (ausencia de elevación de los TG y disminución de AGL plasmáticos)85. Un reciente trabajo evidenció que el tratamiento con exenatide (análogo de GLP-1) durante 4 semanas en hombres sanos redujo la TP-apoB-48 sin modificación de la TP-apoB-100, lo cual destaca al intestino como sitio de acción del GLP-1 en el metabolismo lipídico86 (tabla 1).
El uso de vildagliptina (inhibidor de DPP-IV) durante 4 semanas mejoró la lipemia posprandial (disminución de TG plasmáticos, de TG de quilomicrones y de LRT intestinales) en los pacientes diabéticos tipo 287 (tabla 2). El tratamiento con sitagliptina durante 6 semanas disminuyó las concentraciones de apoB-48 en estado posprandial asociado a una mejoría de la sensibilidad a la insulina en pacientes diabéticos tipo 288 (tabla 2). Además, el uso de exenatide durante 2 semanas en los pacientes diabéticos tipo 2 disminuyó la secreción posprandial de TG89.
Al contrario, el GLP-2 que es secretado en conjunto con GLP-1 exacerba el lipemia posprandial. Se encuentra elevado en el modelo de rata diabética por estreptozotocina, y estaría implicado en la hiperplasia intestinal que acompaña a la diabetes90. Más allá de sus efectos intestinotróficos, la administración aguda de GLP-2 en el caso del hombre estimula la lipemia posprandial probablemente aumentando la absorción intestinal de los lípidos91. Datos recientes en el hámster y el ratón confirman el papel del GLP-2 en la hiperlipemia posprandial (aumento del ensamblaje y de la secreción del LRT intestinales) por aumento de la absorción de los lípidos vía la expresión de la forma glicosilada de CD36/fatty acid translocase a nivel de la membrana apical del enterocito92(fig. 2).
InflamaciónLa RI se asocia con un estado inflamatorio crónico. Sin embargo, pocos estudios se interesaron en el nexo entre la inflamación y la RI a nivel del enterocito. En el modelo de Syrian golden hámster, se demostró que la inflamación inducida por la perfusión del factor de necrosis tumoral alfa (TNF-α) induce una RI intestinal reduciendo la cascada de señalización insulínica (fig. 2). El TNF-α disminuye por un lado la fosforilación de la subunidad-α del receptor de la insulina, de IRS-1 y la Akt, y por otro lado aumenta en el período posprandial, la vía de MAPK/ERK-1/2. Esta RI intestinal se acompaña de una hiperproducción del LRT intestinales en ayunas y posprandial asociada a mayor expresión de MTP y de transportadores como CD36, sin modificar factores de transcripción que participan a la lipogénesis de novo como es el SREBP-1c93.
Estudios de intervenciónVarios trabajos de intervención nutricional o farmacológica han sido realizados con el fin de poder reducir la lipemia posprandial. La presente revisión se focalizará únicamente en los ensayos clínicos que evaluaron el efecto del tratamiento sobre la producción intestinal del LRT. En general, estos estudios comprenden un tamaño de muestra pequeño y un poder estadístico modesto.
Efecto de ácidos grasos libres circulantesEn un modelo animal de síndrome metabólico (gerbille Psammomys obesus), una alimentación rica en ácidos grasos n-3 (omega-3) reduce la síntesis de apoB-48, y el ensamblaje y la secreción del LRT intestinales94. La adición de un tratamiento con ácidos grasos omega-3 (4g/día) a un régimen hipocalórico y un tratamiento con estatina (fluvastatina 80mg/día) redujo significativamente la concentración de apoB-48 en ayunas en un grupo de 8 pacientes diabéticos tipo 2 portadores de hiperlipemia mixta95.
Efecto de las estatinas, los fibratos y el ezetimibeUn tratamiento con estatinas (atorvastatina 20mg/día, durante 6 semanas) redujo la producción intestinal de LRT en un grupo de 6 pacientes diabéticos tipo 2 hipertrigliceridémicos. La misma tendencia, pero no significativa, se observó en el tratamiento con fibrato (fenofibrato 200mg/ día, durante 6 semanas). En cambio, el tratamiento con fenofibrato aumentó el catabolismo del TRL-apoB48, lo que no sucede con la atorvastatina96 (tabla 2). El tratamiento con atorvastatina (10-80mg/ día durante 16 semanas) redujo la concentración de LRT-apoB-48 por estimulación del aclaramiento en estado posprandial en pacientes con HPFC97 (tabla 2). El uso de fenofibrato (200mg/ día durante 6 meses) disminuyó las concentraciones de remanentes de LRT-intestinales en pacientes con HPFC98.
En un grupo de 8 pacientes hipercolesterolémicos, un tratamiento de 8 semanas con ezetimibe no modificó el metabolismo del LRT intestinales99. En cambio, cuando se asoció el tratamiento con ezetimibe a simvastatina (40mg día durante 6 semanas) en un grupo de 16 pacientes que presentaban hiperlipemia mixta, se obtuvo una reducción significativa de la producción del LRT intestinales; esta reducción no se obtiene con simvastatina o ezetimibe solo100.
Evaluación de lipoproteínas ricas en triglicéridos de origen intestinal en la práctica clínicaLa sobreproducción intestinal de LRT constituye un nuevo componente de la RI, participando tanto en el estado posprandial como en estado postabsortivo del aumento de TG plasmáticos. El enterocito es resistente a la acción supresora de la insulina sobre la producción de QM en la diabetes tipo 2 y en la obesidad 40,58. La evaluación de la lipemia posprandial en estados de RI tendría que constituir un elemento más del examen de rutina, intentado detectar hiperlipidemias subclínicas mas marcadas en estados posprandiales que en ayunas. La estandarización de la cantidad de grasa necesaria para estimular el metabolismo de QM continúa siendo una controversia42. En un reciente metaanálisis realizado de 113 ensayos clínicos de lipemia posprandial, proponen la utilización de un nuevo índice, llamado índice lipémico (IL), que consiste en 50-70g de grasa por comida con una evaluación de la lipemia posprandial a través del ABC a las 4 h de finalizada la SOL101,102.
El análisis de apoB-48 en ayunas y en estado posprandial debería formar parte de los exámenes anuales de los estados con RI como diabetes tipo 2, obesidad, HPFC y también enfermedad coronaria, debido a su asociación con la aterosclerosis59,103. La apoB-48 serviría, además, como seguimiento en los tratamientos con estatinas, fibratos y omega-3 95,97. La utilización de la apoB-48 en el área clínica y de investigación son más que evidentes.
ConclusionesNumerosos argumentos indican que el intestino no es un órgano pasivo, sino un órgano metabólicamente activo, recibiendo informaciones de la periferia y siendo capaz de modular sus procesos de síntesis y secreción de lípidos en relación a los sustratos, las hormonas y otras sustancias endógenas o exógenas. Existen parentescos funcionales entre el intestino y el hígado. El papel directo y/o indirecto de la RI enterocitaria, así como los mecanismos celulares y moleculares de acción de estos diferentes factores de regulación permanecen aún sin dilucidar. El análisis de la apoB-48 como indicador de lipemia posprandial y de ateromatosis debería emplearse en la práctica clínica de endocrinología.
El potencial aterogénico del quilomicrón hace necesaria la comprensión de su hiperproducción intestinal y de su acumulación plasmática en la diabetes tipo 2, así como en los síndromes de RI.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores agradecen al Dr. León Litwak su valiosa lectura y correcciones a este trabajo.
