


INTRODUCCIÓN
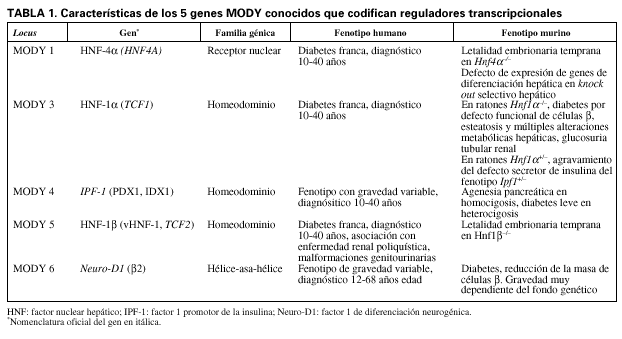
Hace más de 70 años se describió una forma de diabetes que aparece en personas jóvenes y se hereda con un patrón autosómico dominante1. Varias décadas más tarde, Fajans y Tattersall acuñaron el termino maturity onset diabetes of the young (MODY) y progresivamente definieron de un modo más específico este subtipo de diabetes de acuerdo con los siguientes criterios: aparición antes de los 25 años en, al menos, un sujeto, herencia autosómica dominante con penetrancia elevada e, inicialmente, sin evidencia de déficit grave de insulina2,3. En los años noventa, grupos de genetistas humanos reunieron familias que segregaban MODY e identificaron los principales genes que albergaban mutaciones en los familiares afectados. Ello reveló que MODY es en realidad un defecto heterogéneo, tanto desde el punto de vista molecular como clínico (tabla 1). El primer gen implicado en MODY fue el que codifica la enzima glucolítico glucocinasa4,5. Este subtipo de MODY cursa con hiperglucemia leve (en muchos casos sin alcanzar criterios diagnósticos de diabetes mellitus) y condiciona una predisposición a la diabetes gestacional6. Los grupos de Bell y Froguel, por otra parte, determinaron 2 loci genéticos que segregaban con una forma mucho más grave de diabetes en familias MODY, denominados MODY 1 y 37,8. Posteriormente, se realizó un análisis profundo de los genes localizados en estas 2 regiones genómicas, lo que permitió determinar que los genes que albergan las mutaciones causales corresponden a los factores nucleares hepáticos 4* y 1* (HNF-4*y HNF-1*), respectivamente9,10. Este hallazgo fue sorprendente, dado que, a diferencia de la glucocinasa, HNF-4* y HNF-1* no se habían considerado genes candidatos para la diabetes. Existía un gran volumen de información disponible que apuntaba a que la principal función de ambas proteínas consistía en regular la transcripción específica de genes hepáticos11. Sin embargo, el fenotipo de MODY 1 y 3 consiste en una disminución de la secreción de insulina inducida por gluco sa12,13, lo cual claramente indicó que HNF-4* y HNF-1* tienen un papel previamente no sospechado en la función de las células ß. Poco después se identificaron otros 3 reguladores transcripcionales implicados en la diabetes tipo MODY: el factor 1 promotor de la insulina (IPF-1; PDX1/IDX1), HNF-1ß y el factor 1 de diferenciación neurogénica (Neuro-D1/ß2)14-16(tabla 1).

La identificación de los genes implicados en MODY representa un avance muy importante, pero esta información debe utilizarse ahora para comprender la función de estos reguladores transcripcionales en las células ß, e intentar traducir estos descubrimientos en beneficios clínicos. En este artículo se intentarán resumir conocimientos acerca de los factores de transcripción implicados en MODY. En particular se expondrán resultados recientes que han iniciado el camino de la comprensión de los mecanismos implicados en el control de la expresión génica en células ß y sus precursores por parte de estos genes.
EL FACTOR NUCLEAR HEPÁTICO 1* (HNF-1*/TCF1, MODY 3)
Genética molecular de HNF-1*/TCF1 (MODY 3)
HNF-1* es una proteína perteneciente a la familia homeodominio, la cual está compuesta por un número muy diver so de proteínas reguladoras de la transcripción que poseen un dominio unión al ADN con una estructura característica17. HNF-1* se expresa no sólo en el hígado (como parecería indicar su nombre), sino también en el riñón, intestino y páncreas11-18. En el páncreas HNF-1* se expresa desde su formación embrionaria inicial a concentraciones bajas, pero a medida que aparecen células endocrinas y exocrinas diferenciadas las concentraciones se incrementan en ambos tipos celulares de un modo muy notable18.
HNF-1* existe en forma de homodímeros, o bien como heterodímeros HNF-1*/HNF-1ß11. Ambos dímeros forman un complejo con 2 moléculas de una proteína denominada dimerization cofactor of HNF-1 (DCOH). Este complejo tetramérico reconoce secuencias de ADN que se asemejan al motivo palindrómico CGTTAATNATTAACG, típicamente situadas en la proximidad del inicio de transcripción de los genes regulados por HNF-111. La unión al gen diana permite que HNF-1* induzca la activación de su transcripción.
Las mutaciones en el gen de HNF-1*explican la mayor parte de los casos de diabetes MODY, particularmente si excluimos el fenotipo de hiperglucemia leve glucocinasa19. Actualmente se han identificado más de 100 mutaciones del gen de HNF-1*ligadas a MODY19-28. Aproximadamente, la tercera parte está representada por deleciones, inserciones o sustituciones de nucleótidos que interrumpen o modifican el marco de lectura creando una proteína aberrante. Los restantes dos tercios de los casos se deben a sustituciones de nucleótidos que modifican un aminoácido crítico de la proteína (mutaciones missense, o con sentido alterado). Este conjunto de mutaciones es muy informativo, ya que señala residuos de la proteína que son esenciales para la función de HNF-1*. La mayor aglomeración de mutaciones sin sentido se sitúa en la mitad N-terminal de la proteína, en el dominio de dimerización y, particularmente, en el homeodominio utilizado por la proteína para interaccionar y reconocer las secuencias de ADN en sus promotores diana. Este tipo de información se ha utilizado recientemente para resolver la estructura del dominio de unión al ADN de HNF-1*, el cual posee (junto con la región homóloga de HNF-1ß) propiedades singulares en comparación con el conjunto de miembros de la familia de proteínas homeodominio29. También ha sido posible extraer información valiosa a partir de mutaciones humanas situadas en la región promotora del gen de HNF-1*, encargada de su regulación transcripcional. De este modo, una mutación del promotor de HNF-1* que destruye el lugar de unión de HNF-4* y causa MODY demuestra que HNF-4* es necesario para la expresión de HNF-1* en las células ß pancreáticas humanas28.
Fisiopatología del defecto HNF-1*/TCF1 (MODY 3)
La caracterización fenotípica de MODY 3 ha demostrado de forma muy convincente que estos individuos desarrollan diabetes mellitus debido a una incapacidad para segregar insulina cuando se elevan las concentraciones de glucosa8,13. No parece existir resistencia a la acción de la insulina, ni una alteración primaria del metabolismo glucídico hepático30. Asimismo, el análisis de ratones en los que se ha inactivado el gen Hnf1* ha indicado una ausencia de respuesta secretora de insulina al modificar las concentraciones de glucosa, probablemente debida a un defecto en la metabolización de glucosa en las células ß31,32. No obstante, sí existe respuesta secretora a las sulfonilureas, lo cual indica que el defecto de las células ß es específico para ciertas funciones31. Si bien se sabe que la ausencia de Hnf1* en islotes pancreáticos condiciona la expresión defectuosa del transportador de la glucosa GLUT-2, piruvatocinasa y otros genes metabólicos, probablemente el fenotipo de ausencia de respuesta a la glucosa no se debe a la expresión defectuosa de un único gen diana de HNF-1*33. Estudios de expresión con chips de oligonuleótidos Affymetrix realizados en nuestro laboratorio indican que existe un número elevado de genes pancreáticos acinares y endocrinos que se expresan defectuosamente en ratones Hnf1*/ (datos no publicados). Estos genes codifican funciones muy diversas, incluyendo el control del metabolismo de aminoácidos y glúcidos, el control del ciclo celular o diversas vías de transducción de señales. Esto induce a pensar que el fenotipo resultante de la función defectuosa de HNF-1* en las células ß podría ser consecuencia de un fracaso de múltiples funciones celulares. Este hecho no es sorprendente si se considera que antes del experimento con chips de ADN ya se sabía que HNF-1*regula a otros reguladores transcripcionales clave de células ß, cada uno de los cuales regula a múltiples genes diana34,35. Otro hallazgo significativo derivado del análisis de ratones Hnf1*/ es que este factor de transcripción desempeña funciones muy específicas en los islotes pancreáticos, de modo que genes como GLUT-2 requieren Hnf1* en islotes pero no en otros tejidos como el hígado. El estudio del ratón knock-out también ha aportado información acerca del mecanismo de activación de la transcripción dependiente de Hnf1*. Sabemos, por ejemplo, que la activación génica inducida por este regulador está ligada a modificaciones postraduccionales de las colas de histona en los nucleosomas de sus promotores diana33.
Con respecto al fenotipo humano resultante del déficit de HNF-1*, a pesar de que el defecto secretor de insulina en pacientes con MODY 3 puede ser muy importante, no parece deberse a una desaparición completa de las células ß ni a una incapacidad absoluta para transcribir el gen de la insulina. La mejor evidencia de ello es que los sujetos con MODY 3 pueden responder a agentes como las sulfonilureas, que actúan en el canal de postasio dependiente de la adenosintrifosfato y, por lo tanto, distalmente al mecanismo sensor de la glucosa36. Además del significado fisiopatológico, esta observación tiene implicaciones terapéuticas obvias para pacientes con MODY 3.
Existen ya abundantes pruebas de que la aparición de MODY 3 sobreviene por un mecanismo de haploinsuficiencia. Este término hace referencia a que el fenotipo es una consecuencia de la simple pérdida de función de uno de los 2 alelos, no de un efecto dominante negativo, en el que la mutación en un alelo inhibe la función del alelo sano. Probablemente la mejor prueba de ello es la observación previamente mencionada de la mutación en el promotor de HNF-1*, la cual es difícilmente compatible con un efecto dominante negativo28. Otras evidencias que apoyan este concepto se basan en estudios de caracterización funcional de las mutaciones MODY 3, que frecuentemente demuestran una pérdida de función, y la observación de que el alelo mutado en ocasiones no se expresa debido a una degradación selectiva del ARN por el mecanismo de nonsense mediated decay21,37.
Una propiedad interesante del defecto en MODY 3 que aún no se ha explicado de forma satisfactoria es que no está presente al nacimiento, sino que surge años más tarde. De hecho, raramente se observa en la primera década de la vida19,38. A partir de la segunda década la incidencia crece de modo abrupto, de modo que a los 40 años la penetrancia es casi completa, si bien la gravedad puede ser muy diversa incluso dentro de una misma genealogía19. Aunque es tentador pensar que factores como la obesidad o el sedentarismo pueden estar implicados en la heterogeneidad de la gravedad clínica o en la edad de aparición, esto no se ha demostrado. Es muy interesante señalar que, hasta la fecha, los estudios realizados no han identificado una relación entre la gravedad del fenotipo y la naturaleza de la mutación19. Una interpretación posible de este hecho, de ser confirmado en estudios futuros más extensos, es que las mutaciones sólo causan MODY si producen una pérdida de función completa o casi completa. La penetrancia en estas circunstancias estaría condicionada por factores ambientales o relacionados con la historia genética del individuo.
EL FACTOR NUCLEAR HEPÁTICO 4* (HNF-4*/HNF-4A, MODY 1)
La diabetes autosómica dominante de aparición temprana producida por mutaciones en el gen que codifica HNF-4* es mucho menos frecuente que MODY 39. Clínicamente el fenotipo es casi indistinguible de la diabetes HNF-1*, a excepción de que posiblemente existe una reducción relativa en las concentraciones de apolipoproteínas AII y CIII, así como de triglicéridos, mientras que en MODY 3 existe una disminución de apolipoproteína M39,40. Además, a diferencia de la diabetes HNF-1*, no existe una reducción en el umbral de glucosuria41. Al igual que ocurre en MODY 3, el defecto en MODY 1 parece obedecer a un mecanismo de haploinsuficiencia42-44.
HNF-4* es un miembro de la familia de receptores nucleares que, al igual que HNF-1*, se expresa en el hígado, riñón, intestino y páncreas11. Muchos receptores nucleares de esta familia están sujetos a regulación por hormonas o metabolitos, pero hasta ahora no se ha descrito de forma concluyente un ligando para HNF-4*, por lo que sigue considerándose un receptor "huérfano". Recientemente, se ha establecido la estructura de HNF-4* y se ha constatado que el dominio de unión al ligando está ocupado por ácidos grasos, pero éstos podrían corresponder a un componente constitutivo de la estructura de HNF-4* más que a una molécula capaz de regular de forma aguda la función de HNF-4*45,46. No obstante, independientemente de una hipotética función fisiológica de estos ácidos grasos, constituyen una diana atractiva para crear ligandos artificiales capaces de modular la función de HNF-4* farmacológicamente.
La información sobre el papel de HNF-4* en el páncreas aportada por los estudios animales ha sido mucho más pobre que en el caso de HNF-1*, puesto que los ratones con mutaciones homocigotas nulas de Hnf4* tienen un defecto embrionario temprano letal47. No obstante, existen varios motivos para pensar que los defectos moleculares son similares en ambos casos. Además de la similitud del fenotipo humano, la inhibición de HNF-1* o HNF-4* en células ß tumorales da lugar a un patrón de expresión génica extraordinariamente parecido48,49. Por otra parte, varios estudios han demostrado que HNF-4* controla la expresión de HNF-1* en células hepáticas y endodermo, y existe evidencia genética humana, mencionada anteriormente, que apoya que esto es así en los islotes pancreáticos28,50-52. En ratones HNF-1* es, a su vez, necesario para la expresión de HNF-4*, aunque esto ocurre sólo en los islotes pancreáticos. Esto se debe a que en los islotes pancreáticos existe un promotor alternatio de HNF-4*, denominado P2, que confiere dependencia a HNF-1*34,44. Esta dependencia se da también en humanos, ya que una mutación en el promotor P2 que reduce la afinidad de HNF-1* por su secuencia diana produce diabetes en una familia con MODY 142. Esta circunstancia indica que existe interdependencia entre ambos factores selectivamente en los islotes pancreáticos, donde forman un circuito de regulación cruzada específico de este tipo celular53. Este circuito genético tiene implicaciones interesantes en la fisiopatología de MODY, tal como se comentará más adelante.
FACTOR 1 PROMOTOR DE LA INSULINA (IPF-1/IDX1/PDX1, MODY 4)
El gen IPF-1, que codifica la proteína homeodominio conocida como PDX1 o IDX1, es necesario para la progresión del rudimento pancreático embrionario54,55. Los ratones Ipf1/ tienen agenesia pancreática, y se ha descrito un humano con agenesia pancreática por una mutación nula homocigota de IPF-156,57. El análisis genealógico extendido de este caso índice con agenesia demostró que la misma mutación en estado heterocigoto cosegrega con diabetes de aparición temprana siguiendo un patrón autosómico dominante, conocido como MODY 415. Más recientemente se ha identificado otra mutación del gen IPF-1 que cosegrega con diabetes siguiendo un patrón consistente con MODY58. Varios estudios han confirmado que la heterocigosidad del gen IPF-1 también da lugar a diabetes en ratones, lo que parece deberse a que existe un defecto en la secreción de insulina inducida por glucosa, posiblemente debido a un defecto en la expresión de SUR1 y KIR6.2 y de moléculas que regulan el procesamiento de insulina59-61. Aunque las mutaciones heterocigotas en IPF-1 no son una causa frecuente de MODY15,58, varios grupos han descrito mutaciones "leves" de IPF-1 (que causan un reducción parcial de su actividad) que podrían predisponer a diabetes tipo 2 de aparición más tardía, sin reunir criterios para definirla como MODY62,63. En resumen, tanto la genética de ratones como los estudios humanos indican que IPF-1/PDX1 tiene mútliples funciones en diferentes momentos del desarrollo: inicialmente es necesario para la formación del páncreas y, posteriormente, es requerido para mantener sus funciones diferenciadas endocrinas.
EL FACTOR NUCLEAR HEPÁTICO 1ß (HNF-1ß/TCF2/VHNF-1, MODY 5)
HNF-1ß es una proteína estructuralmente muy parecida a HNF-1* en sus dominios de dimerización y unión al ADN, si bien difiere sustancialmente en su región C-terminal11. Se expresa en el páncreas, epitelio genitourinario y de las vías biliares, así como en el pulmón. Tiene un papel importante en el desarrollo endodérmico temprano, de modo que los embriones con mutaciones nulas homocigotas mueren antes de la organogénesis pancreática64. La diabetes por defectos heterocigotos en HNF-1ß es rara, pero se distingue claramente de otras formas de diabetes por la coexistencia de enfermedad renal quística que con frecuencia condiciona insuficiencia renal y malformaciones del tracto genitourinario65,66. La imposibilidad de estudiar el páncreas en animales con mutaciones homocigotas para este gen ha limitado nuestro conocimiento acerca de la función de HNF-1ßen las células ß, por lo que es de esperar que nuevos modelos basados en la inactivación condicional de HNF-1ß aporten mucha más información. Estudios recientes, sin embargo, han demostrado que HNF-1ß tiene funciones muy diferentes de su homólogo HNF-1* y, en consecuencia, que la fisiopatología de la diabetes MODY 3 y MODY 5 difiere sobremanera. De este modo, mientras que HNF-1* está presente predominantemente en las células diferenciadas del páncreas embrionario, HNF-1ß está enriquecido en las células ductales embrionarias y adultas, pero no es detectable en las células productoras de insulina18. Un análisis detallado de las células embrionarias que expresan HNF-1ß indica que éstas constituyen la población de células precursoras multipotentes que dan lugar a los linajes endocrino o ductal exocrino diferenciados18. Esto apunta a que, a diferencia de HNF-1*, HNF-1ß podría ser un regulador de la génesis de células endocrinas y exocrinas, y no un regulador de la función de las células ß diferenciadas. En concordancia con este concepto, los pacientes con MODY 5 presentan hipoplasia pancreática grave67.
HNF-6, un regulador transcripcional homeodominio que es indispensable para la formación de células endocrinas durante la embriogénesis pancreática, es, a su vez, necesario para que se active el gen de HNF-1ß en las células precursoras pancreáticas. Ello indica que ambos genes, HNF-1ß y HNF-6, forman una red de regulación transcripcional que controla la función de las células precursoras embrionarias pancreáticas, si bien queda por resolver de qué modo activan programas relacionados con la especificación de los diferentes linajes.
EL FACTOR HÉLICE-ASA-HÉLICE NEURO-D1/ß2 (MODY 6)
Neuro-D1 (también llamado ß2) es un regulador transcripcional de la familia basic helix-loop-helix que clonaron 2 laboratorios basándose en su capacidad de unirse y activar el gen de la insulina, o bien de inducir neurogénesis68,69. Se han descrito 3 mutaciones en este gen que causan MODY, si bien la caracterización fenotípica es más limitada en comparación con otras formas de MODY16,70. Los ratones con mutaciones homocigotas de este gen desarrollan diabetes, en gran parte por una reducción de la masa de células ß por apoptosis, con desestructuración de los islotes68,71. Es interesante el hecho de que, dependiendo del fondo genético, la diabetes puede revertirse con el tiempo por neoformación compensatoria de células ß71. No se ha publicado hasta la fecha un análisis detallado de la función de las células ß en ratones heterocigotos.
¿POR QUÉ EXISTE UN FENOTIPO ESPECÍFICO EN CÉLULAS ß CUANDO EXISTE UNA MUTACIÓN DE UN SOLO ALELO DE LOS GENES MODY?
El hecho de que mutaciones en un solo alelo de algunos genes de reguladores transcripcionales origine diabetes tipo MODY plantea una paradoja importante. Los ratones con inactivación de ambos alelos de Hnf1* presentan una disfunción hepática y tubular grave, además de diabetes72,73. Los ratones Hnf4*/ no sobreviven más allá del estado embrionario E6, y la inactivación complta del gen HNF-1ß también da lugar a letalidad embrionaria temprana64. ¿Por qué la pérdida de un solo alelo en cualquiera de estos 3 genes genera un defecto relativamente específico de las células ß? Evidentemente, debe de existir una función de estos reguladores transcripcionales que es específica de las células ß, y singularmente vulnerable a la dosis génica. Ello podría estar vinculado a la interdependencia entre HNF-1* y HNF-4* selectivamente en las células ß53. La presencia de un solo alelo puede reducir la concentración del producto del gen mutado y, además, reducir la capacidad de compensar fluctuaciones de expresión que normalmente se producen a partir de cada alelo74. Esto puede favorecer que una reducción del producto del gen mutado alcance el nivel umbral para reducir suficientemente la expresión del gen opuesto, lo que desencadenaría una extinción duradera de la expresión de ambos. La acumulación de células ß que han sufrido esta transición al cabo de varios años puede alcanzar el nivel requerido para dar lugar a diabetes mellitus. Este tipo de fenómeno sólo es de esperar que ocurra en las células ß, puesto que en otras células no existe interdependencia.
IMPLICACIONES TERAPÉUTICAS DE LOS CONOCIMIENTOS ACERCA DE LA GENÉTICA DE MODY
El descubrimiento de la importancia de los reguladores transcripcionales MODY en las células ß ha supuesto un gran salto cualitativo en nuestra comprensión del fenotipo diferenciado de estas células. El hecho de que los individuos con mutaciones heterocigotas en estos genes tengan un fracaso de la función de las células ß aporta evidencia inequívoca de la trascendencia de dichos genes, y tiene implicaciones que van más allá de la enfermedad MODY per se. Uno de los aspectos más llamativos de los primeros estudios encaminados a entender el fenotipo MODY estriba en que existe una gran interconexión funcional entre los diferentes reguladores transcripcionales implicados. Ello explica la similitud del fenotipo resultante cuando se producen mutaciones en diferentes genes. Además, apunta hacia la existencia de un sistema regulador complejo previamente no sospechado que, en su conjunto, desempeña un papel crítico en la función de las células productoras de insulina.
En los últimos años la diferenciación artificial de las células ß para su utilización como tratamiento celular sustitutivo de la diabetes tipo 1 se ha convertido en una opción extremadamente prometedora75. Este tipo de célula artificial deberá contar no sólo con la capacidad de producir y segregar suficiente insulina, sino también con un sistema regulador transcripcional HNF-1*/HNF-4*intacto. Asimismo, el hallazgo de que el gen MODY 5 (HNF-1ß) forma parte de una red reguladora de las células precursoras pancreáticas adquiere una trascendencia obvia en cualquier intento de forzar el proceso de diferenciación hacia la célula productora de insulina18.
Por otra parte, las descripciones de mutaciones "leves" en HNF-1* e IPF-1 asociadas a diabetes tipo 2 de aparición tardía indican que debe explorarse en qué medida algunos de los componentes de la red de control transcripcional pueden estar implicados en la fisiopatología de formas de diabetes sin las características de MODY62,63,76. Por último, es de esperar que la disponibilidad de una subcategorización molecular de nuevas formas de diabetes humana y la existencia de modelos genéticos murinos claramente relevantes y similares al fenotipo humano permita validar la efectividad de agentes terapéuticos específicos para pacientes con distintas formas de MODY.
