


El descubrimiento de la existencia de hormonas gastrointestinales que modulan la homeostasis energética ha despertado un gran interés. Algunas de estas hormonas, actuando en el hipotálamo o el núcleo del tracto solitario en el tronco encefálico, ejercen efectos moduladores del apetito y la saciedad. En términos generales, las señales endocrinas generadas en el tracto gastrointestinal tienen efecto anorexigénico directo o indirecto a través del sistema nervioso vegetativo. Sólo la ghrelina, hormona producida en el estómago, se ha asociado de manera consistente con el inicio de la ingesta y se la considera una de las principales señales orexigénicas en los modelos animales estudiados y en humanos. En esta revisión, se describen brevemente las principales hormonas de origen gastrointestinal implicadas en la regulación del apetito. Dada la importancia que los trastornos de la ingesta de alimentos, especialmente la obesidad, han adquirido, un mejor conocimiento de los mecanismos de acción de estas señales endocrinas podría contribuir al desarrollo de nuevas moléculas que incrementen y mejoren nuestro arsenal terapéutico para tratar la obesidad y las enfermedades crónicas relacionadas con ella.
The discovery of gut hormones regulating the energy balance has aroused great interest in the scientific community. Some of these hormones modulate appetite and satiety, acting on the hypothalamus or the solitary tract nucleus in the brainstem. In general, the endocrine signals generated in the gut have direct or indirect (through the autonomous nervous system) anorexigenic effects. Only ghrelin, a gastric hormone, has been consistently associated with the initiation of food intake and is regarded as the main orexigenic signal both in animal models and humans. In this review, we provide a brief description of the major gastrointestinal hormones implicated in the regulation of food intake. Given the increased importance of food intake disturbances, especially obesity, a better understanding of the underlying mechanisms of action of the gastrointestinal hormones might contribute to the development of new molecules that could increase the therapeutic arsenal for treating obesity and its associated comorbidities.
La ingesta de alimentos es una actividad imprescindible para la vida y, por ello, está bajo el control de una serie de mecanismos sofisticados que interaccionan entre sí para conseguir un balance energético adecuado. Sin embargo, cuando alguno de estos mecanismos falla y el equilibrio se rompe, pueden generarse alteraciones fisiopatológicas como la obesidad.
La obesidad es uno de los principales problemas de las sociedades desarrolladas, no sólo por la afección en sí, sino por las numerosas complicaciones que suele conllevar: diabetes mellitus, enfermedades cardiovasculares, cáncer, hipertensión arterial, etc. Por todo ello, es de gran importancia conocer todos los mecanismos que participan en el control del apetito y la homeostasis energética, lo que puede contribuir a prevenir y tratar tanto la obesidad como las enfermedades crónicas relacionadas.
El control del peso corporal ha suscitado gran interés en el entorno científico desde siempre. En un principio se pensó que el control del apetito era un proceso regulado exclusivamente en el sistema nervioso central. A partir de experimentos en los que se lesionaban o estimulaban distintas regiones cerebrales, se demostró la importancia en la ingesta de ciertos núcleos hipotalámicos, como el hipotálamo lateral o el núcleo ventromedial. Así se estableció la teoría dual, que postulaba la existencia de un centro del hambre y un centro de la saciedad. Más adelante se descubrió que en este proceso estaban implicados otros núcleos y regiones cerebrales extrahipotalámicas1. Con el tiempo, se determinó que la regulación de la ingesta no era un proceso exclusivo del sistema nervioso central, sino que en él también participaban señales periféricas. Así, se demostró la importancia de la leptina y el tejido adiposo en lo que se conoce como regulación a largo plazo, la influencia del eje adrenal en la ingestión de alimentos o la importancia de señales procedentes del páncreas o del tracto gastrointestinal en la regulación de la ingesta a corto plazo2. Estas señales periféricas llegan al sistema nervioso central por vía neuronal (a través del nervio vago) o por vía humoral (como secreciones endocrinas que se vierten al torrente sanguíneo). Pero en el ser humano también adquieren gran importancia en los patrones de ingesta otros elementos como el estilo de vida (cada vez más sedentario), la conducta social y factores hedónicos relacionados con el alimento. Recientemente, también se ha demostrado la existencia de una programación fetal o neonatal3.
En esta revisión pretendemos hacer una recapitulación de las principales señales periféricas de origen gastrointestinal y analizar su posible participación en el control de la ingestión de alimentos.
Desde que Starling y Bayliss (1902) identificaron la secretina como molécula encargada de las secreciones pancreáticas, sabemos que el tracto gastrointestinal (GI) no sólo es un sistema donde se almacenan y se procesan los alimentos, sino que también es determinante en su propia regulación, en el control de la ingesta y en el balance energético. Sin embargo, no ha sido hasta tiempos más recientes cuando se han identificado las señales que participan en esta regulación. Existen numerosos péptidos que son liberados desde el estómago o el intestino y participan en las sensaciones de saciedad o de hambre, que por vía neuronal o humoral van a originar la activación de diferentes regiones cerebrales, induciendo la ingesta o su cese. Se establece así una conexión recíproca entre cerebro y tracto GI. Estos péptidos de origen GI son en su mayoría señales de saciedad, y su descubrimiento ha supuesto un enorme avance científico en el campo del desarrollo de fármacos antiobesidad.
HORMONAS ANOREXIGÉNICASLa mayoría de las hormonas de origen gastrointestinal que intervienen en el control de la ingestión de alimentos pertenecen a este grupo. El papel de algunas de ellas como moléculas saciantes se conoce desde hace tiempo, pero la relevancia fisiológica de cada una y de todas en su conjunto está por establecer.
Colecistocinina (CCK)Se sintetiza principalmente en las células L del duodeno y el yeyuno, aunque también se libera como neurotransmisor en varias regiones cerebrales (córtex, amígdala, hipocampo, tálamo e hipotálamo, entre otras)4. La ingestión de nutrientes, fundamentalmente grasas y proteínas, induce la liberación de CCK al torrente sanguíneo. Se han descrito varias formas moleculares que difieren en el número de aminoácidos5: CCK-8, CCK-33, CCK-39 y CCK-58.
Sus efectos biológicos están mediados por dos tipos de receptores acoplados a proteínas G, el CCK1 y el CCK2, en un inicio denominados CCKa y CCKb, respectivamente6.
Primeramente, se describió su capacidad de contraer la vesícula biliar. Después se le atribuyeron otras funciones gastrointestinales como la estimulación de la secreción de enzimas pancreáticas o la inhibición del vaciamiento gástrico4.
En varias especies, incluido el hombre, la administración periférica de CCK causa saciedad y disminuye la cantidad de alimento ingerido en cada período de ingesta7-9. Esta respuesta anorexigénica es dependiente de la dosis de CCK administrada y parece estar mediada por los receptores CCK1 situados en las fibras aferentes del nervio vago, que van a transmitir la señal al núcleo del tracto solitario (NTS) para producir sensación de saciedad10. La vagotomía subdiafragmática evita el efecto saciante de la CCK11,12. Esta inhibición de la ingesta también se observa en ratas tras la administración de CCK exógena directamente en el sistema nervioso central. En las ratas Otsuka Long-Evans Tokushima fatthy (OLETF), que presentan una mutación espontánea del receptor CCK1 y un fenotipo obeso, se ha observado un incremento de la ingesta13,14.
La infusión intravenosa de CCK-33 en seres humanos causó un descenso en el volumen de la ingesta y redujo la sensación de hambre posprandial15. El empleo de antagonistas del CCK1, como loxiglumida, en seres humanos sanos inhibe el efecto saciante de la administración intraduodenal de grasas y bloquea la reducción del apetito inducida por la administración preprandial de CCK-816. Sin embargo, el empleo de agonistas de CCK como la ceruleína no consiguió reproducir estos resultados. La administración de CCK en ratas delgadas o en ratas obesas Zucker, no modifica la ingesta de alimentos17.
La CCK se considera una señal de saciedad a corto plazo, pero no parece afectar al control del balance energético a largo plazo. Su administración periférica crónica no modifica el peso corporal ya que, para compensar la respuesta anorexigénica inducida por el péptido, se produce un incremento en la frecuencia de las comidas18. Tampoco se observó pérdida de peso en pacientes obesos o con sobrepeso sometidos a un tratamiento prolongado con GI181771, agonista del receptor CCK119. Además, si la CCK se administra continuamente, se desarrolla tolerancia a corto plazo (24h)20. No obstante, su efecto en la ingesta sí que parece estar influido por la insulina y la leptina, principales hormonas de la regulación a largo plazo. Cuando por vía central se administran junto con insulina o leptina, se incrementa la sensibilidad a la CCK y se obtiene como resultado final un efecto sinérgico en la regulación del apetito21-23.
La CCK además interacciona con otras hormonas. Algunos estudios demuestran que la administración periférica de CCK inhibe el efecto orexigénico inducido por la administración intraperitoneal de ghrelina24. Por otra parte, se ha comprobado que el péptido tirosina-tirosina (PYY) inhibe el efecto de la CCK estimulador de la secreción de enzimas pancreáticas25. La CCK y su receptor CCK1 participan en la inhibición de la secreción de ghrelina y en la estimulación de la liberación de PYY, inducidas por la administración intraduodenal de grasas. Ambos procesos se pierden tras la administración de dexloxiglumida, antagonista del receptor CCK126.
Los individuos obesos presentan unos valores de CCK circulante elevados, mientras que en la anorexia son bajos27. En individuos con síndrome de PraderWilli, la concentración plasmática de CCK es similar a la observada en los sujetos obesos28.
Péptido tirosina-tirosinaEl péptido fue aislado en 1980 de la mucosa del yeyuno de cerdo por Tatemoto et al29. Pertenece a la familia del polipéptido pancreático (PP), que además incluye el neuropéptido Y (NPY), con los que comparte un 70% de homología. Esta familia de péptidos ejerce sus funciones a través de cinco receptores de membrana acoplados a proteínas G (denominados Y1, Y2, Y4, Y5 e Y6), que se clasifican según su distribución y su diferente afinidad por PYY, PP y NPY30.
PYY se sintetiza en las células L del tracto gastrointestinal distal, principalmente colon y recto, pero también está presente en estómago, páncreas y determinadas regiones del sistema nervioso central31.
Inicialmente se libera como PYY (1–36), molécula constituida por 36 aminoácidos que contiene un residuo de tirosina en cada uno de sus extremos, lo que le confiere el nombre de péptido tirosina-tirosina. Sin embargo, de todo el PYY liberado, alrededor del 40% pierde sus dos aminoácidos iniciales por acción de la enzima dipeptidilpeptidasa (DPP) IV y se convierte en PYY (3–36), que es la forma mayoritaria en circulación32.
PYY (1–36) se une a los receptores Y1, Y2, Y4 e Y5; mientras que PYY(3–36) tiene una afinidad más específica por los receptores Y2, distribuidos principalmente en el núcleo arcuato (ARC) hipotalámico, el hipocampo, el intestino y el nervio vago. La unión de PYY a estos receptores desencadena respuestas inhibitorias como la degradación de AMPc30,33.
Su secreción depende de la ingesta calórica y de la composición de los alimentos, pues la estimulan fundamentalmente los lípidos34. Otros factores como las sales biliares, el ácido gástrico, el péptido intestinal vasoactivo (VIP) y la CCK parece que también estimulan su secreción. Sin embargo, la distensión gástrica no la afecta35. Durante el ayuno, los valores de PYY plasmáticos son bajos y se incrementan en los 15–30min del comienzo de cada comida y permanecen elevados durante 6h35. Una vez liberado a la sangre, tiene múltiples efectos. Promueve la absorción de los nutrientes retrasando el vaciamiento gástrico y el tránsito intestinal. Además, en el intestino delgado inhibe la secreción de fluidos y electrolitos36. Su infusión intravenosa tiene efectos en el sistema cardiovascular, donde induce una respuesta vasoconstrictora e incrementa la presión arterial. En los riñones reduce la velocidad de filtración glomerular, disminuye la renina e incrementa la excreción de sodio, con lo que participa en la natriuresis posprandial35.
Los valores de PYY se mantienen bajos antes de las comidas y se produce un incremento posprandial. La administración periférica de PYY (3–36) induce una respuesta hipofágica en roedores tanto sometidos a ayuno como con disponibilidad ad libitum de comida37. Estos mismos resultados se reproducen en otras especies animales38 y en seres humanos sanos, en quienes la infusión por vía intravenosa reduce el apetito y la ingesta de alimentos durante más de 24h39. En humanos obesos también se observa una reducción de la ingesta de hasta el 30% tras la administración intravenosa o subcutánea de PYY (3–36)39. La administración crónica, además de reducir la ingesta, induce un menor incremento de peso corporal en ratones, conejos y macacos Rhesus, por lo que parece participar en la regulación de la homeostasis energética a largo plazo38,40. Este efecto saciante de PYY se confirma en el ratón deficiente en PYY, que ingiere una mayor cantidad de alimento por día y presenta un mayor peso corporal y más cantidad de tejido graso41.
El efecto anorexigénico de PYY (3–36) está mediado fundamentalmente por el nervio vago y se pierde en ratas vagotomizadas42,43. También se ha demostrado la capacidad de PYY (3–36) de atravesar la barrera hematoencefálica, por lo que puede actuar directamente sobre los receptores Y2 presentes en el hipotálamo. Ahí interacciona con las poblaciones neuronales productoras de NPY y de proopiomelanocortina (POMC), por lo que se produce una reducción en el consumo de alimentos.
De este modo, la administración de PYY (3–36) a nivel central genera respuestas antagónicas según la región en la que se inyecte el péptido y dependiendo del tipo de receptor que se active. Su administración directamente en el ARC actúa sobre los receptores Y2 presinápticos, que incrementan la liberación de POMC y reducen la de NPY, lo que desencadena una respuesta hipofágica44. La deleción de dicho receptor en el hipotálamo produce reducción del peso corporal e incremento de la ingesta45. Sin embargo, la inyección del péptido en los ventrículos cerebrales, a diferencia de lo que ocurre tras el tratamiento periférico, estimula el apetito y la ingesta46. Esta respuesta está atenuada en ratones deficientes en Y1 o Y5, por lo que este efecto orexigénico a nivel central parece deberse a la activación de estos receptores47.
La administración de PYY a seres humanos no parece modificar las concentraciones circulantes de insulina, leptina, GLP-1 (glucagon-like peptide-1) o PP; sin embargo, sí que tiene efecto inhibitorio de la ghrelina. También se ha observado que la secreción de PYY en respuesta a la ingestión de grasas depende en gran medida de la CCK35.
En la obesidad, los valores de PYY están reducidos respecto a los sujetos delgados48; sin embargo, no se modifican en pacientes con síndrome de Prader-Willi (PWS)49. En la bulimia se pierde el incremento de los valores circulantes de PYY en respuesta a la ingestión de alimentos, y en hembras adolescentes con anorexia nerviosa no se observaron diferencias entre las cifras basales y posprandiales de PYY respecto a las de los controles sanos. La concentración plasmática de PYY se incrementa tras la cirugía de bypass y podría contribuir a la pérdida de peso observada en estos pacientes50. A pesar de todo, debido a su corta vida media en plasma y a la necesidad de administrarlo por vía parenteral, el empleo de PYY en terapia clínica está limitado.
Polipéptido pancreáticoEn 1975, el grupo de Kimmel consiguió aislar de extractos de páncreas de pollo un péptido de 36 aminoácidos con gran capacidad de inhibir la función exocrina pancreática51. Recibió el nombre de polipéptido pancreático y, junto con PYY o NPY, constituye la familia de péptidos PP. Al igual que lo que ocurre con el resto de los péptidos de esta familia, sus efectos están mediados por los receptores Y, y el PP es más afín por los receptores Y4 e Y530.
Se sintetiza principalmente en una subpoblación de células situadas en la periferia de los islotes de Langerhans del páncreas, que reciben el nombre de células tipo F o células PP, aunque también se ha detectado su expresión en el páncreas exocrino y en regiones distales del tracto gastrointestinal como el colon o el recto31.
El PP es secretado tras la ingestión de alimentos en correlación con el número de calorías ingeridas. Una vez liberado al torrente sanguíneo, la concentración de PP permanece incrementada durante aproximadamente las 6h siguientes a la ingestión52.
La secreción del péptido se ve favorecida por ciertas hormonas pancreáticas y gastrointetinales como ghrelina, secretina o CCK, mientras que la somatostatina y sus análogos la inhiben. Otros factores como la activación adrenérgica, el tono vagal o la distensión gástrica pueden afectar a su secreción52.
En el tracto gastrointestinal, el PP, además de inhibir la función exocrina del páncreas, reduce la contracción de la vesícula biliar, retrasa el vaciamiento gástrico (aunque los estudios son controvertidos) y reduce el tránsito intestinal52.
El PP reduce la ingesta de comida en roedores y humanos delgados cuando se administra por vía periférica53. Este efecto anorexigénico también se observó en pacientes con síndrome de Prader-Willi, en los que la administración de dos inyecciones diarias de PP redujo en un 12% la ingesta de alimentos54. La administración periférica crónica de PP en ratones ob/ob produce un menor incremento de peso y mejora la resistencia a la insulina y la dislipemia55. También se ha comprobado que la sobrexpresión de PP en un ratón transgénico produce hipofagia y determina un fenotipo más delgado que el de sus respectivos controles. La inyección de anticuerpos contra el PP puede revertir este fenotipo delgado56.
El PP no atraviesa la barrera hematoencefálica, pero su administración periférica parece activar neuronas en el área postrema (AP), región en la que dicha barrera es menos selectiva. En este proceso parecen ser de gran importancia los receptores Y4 que abundan en esta zona y en el nervio vago57. De hecho, la vagotomía bloquea el efecto hipofágico de la administración intraperitoneal de PP en roedores58. Además el tratamiento con antagonistas de los receptores muscarínicos reduce la liberación posprandial de PP un 60%59.
Por otro lado, se ha observado que la administración periférica de PP en ratones inhibe la expresión hipotalámica de los neuropéptidos orexigénicos NPY y orexinas, por lo que el PP puede actuar directamente sobre los receptores Y4 que se expresan en el ARC y en el núcleo paraventricular (PVN)55,60.
A este efecto anorexigénico del PP también puede contribuir que el péptido induce la reducción de la concentración plasmática de ghrelina y su expresión en el estómago de ratones y pacientes con PWS54.
Sin embargo, la administración de PP a nivel central tiene un efecto opuesto al de la administración periférica, pues produce incremento de la ingesta y el vaciamiento gástrico61. En este caso, los receptores implicados en el proceso parecen ser los Y5, pues el ratón deficiente en este tipo de receptores manifiesta una respuesta hiperfágica atenuada tras la administración de PP a nivel central62,63.
Los sujetos anoréxicos presentan valores posprandiales de PP más elevados que los individuos sanos64; por el contrario, las cifras posprandiales de PP están reducidas en individuos obesos65, por lo que existe una correlación inversa entre adiposidad y valores plasmáticos de PP. El PP, además, parece estar implicado en la patogenia del síndrome de Prader-Willi. Estos pacientes presentan una respuesta de PP a la ingesta de alimentos atenuada, y tienen cifras basales y posprandiales más bajas que sus controles sanos66.
Por todo ello, el PP podría ser utilizado como supresor del apetito, o sus antagonistas podrían ser útiles en el tratamiento de la anorexia. Sin embargo, todavía se desconoce el papel de PP en la obesidad.
Polipéptido insulinotrópico dependiente de glucosa (GIP)Péptido de 42 aminoácidos descubierto por Brown et al, quienes observaron que la infusión intravenosa de una preparación de péptidos provenientes del intestino inhibía la secreción de ácido gástrico y pepsina, así como la motilidad del antro y del fundus de perros67. El péptido que causa dicho efecto se llamó inicialmente polipéptido inhibidor gástrico. Sin embargo, en seres humanos este efecto es menos importante que el observado en perros. Posteriormente se descubrió la capacidad de GIP de inducir la secreción de insulina en presencia de concentraciones elevadas de glucosa, por lo que se le cambió la denominación por la actual (polipétido insulinotrópico dependiente de glucosa), haciendo referencia a su efecto incretina pero manteniendo las mismas siglas68,69.
El GIP se sintetiza y se libera tras ingerir hidratos de carbono o lípidos fundamentalmente, desde las células K del intestino delgado (principalmente en el duodeno, aunque también en menor cantidad en yeyuno e íleon) y en zonas del sistema nervioso central y en la glándula salival. Al igual que glucagón, GLP, VIP o somatoliberina, entre otros, el GIP pertenece a la superfamilia de la secretina-glucagón68,69.
El péptido maduro consta de 42 aminoácidos (GIP 1-42) y su vida media en plasma es de aproximadamente 7min. Contiene una alanina en la posición 2 del extremo N-terminal que lo convierte en sustrato de la DPP IV, enzima que lo transforma en el fragmento truncado GIP 3-42, que tiene actividades antagónicas a las de la molécula completa70.
El GIP ejerce sus efectos a través de un único receptor descrito, el GIP-R, que presenta una homología del 40 y el 44% con los receptores de GLP-1 y glucagón, respectivamente68,69.
Entre los efectos de GIP, debemos destacar su actividad incretina71,72. Durante el ayuno, las concentraciones de GIP se mantienen bajas. La ingestión de glucosa o grasa desencadena la liberación de GIP y se produce un incremento en su concentración plasmática, hecho que favorece la secreción de GLP-1 desde las células L intestinales. De esta manera, ambas hormonas promueven la secreción de insulina desde las células beta de los islotes de Langerhans, en lo que se conoce como mecanismo incretina. Sin embargo, se ha comprobado que el efecto insulinotrópico de la administración de GIP en pacientes con diabetes mellitus tipo 2 es menos eficiente que en los individuos sanos, por lo que en este caso la secreción posprandial de insulina no es suficiente para normalizar la glucemia73. A pesar de que los valores de GIP están elevados, parece que en los individuos diabéticos se produce una reducción de la sensibilidad, por una expresión deficiente del receptor en las células beta del páncreas74.
Además de estimular la secreción de insulina, el GIP ejerce efectos antiapoptóticos e induce el crecimiento de las células beta en el páncreas. En el hueso tiene efectos anabólicos y está implicado en la proliferación neuronal en el sistema nervioso central, donde también puede influir en aspectos relacionados con el aprendizaje y la memoria68,69.
Los estudios sobre la influencia de GIP en la ingesta de alimentos son escasos y algo controvertidos. Verdich et al señalan la participación de GIP en la regulación del apetito en humanos75. Sin embargo, en otro estudio se observa que la glucosa y la fructosa son igualmente efectivas suprimiendo la ingesta, a pesar de las grandes diferencias observadas en la secreción de GIP en respuesta a la administración de cada sacárido76. Recientemente, se ha descrito que la infusión de GIP en individuos sanos de peso normal puede reducir el gasto energético y la sensación de hambre; sin embargo, esto no ocurre en pacientes con diabetes mellitus tipo 277.
A pesar de su escaso efecto sobre el control de la ingesta, se ha relacionado a GIP con la obesidad. En los adipocitos, se ha detectado una amplia presencia de receptores de GIP78 y se sabe que en el tejido adiposo GIP estimula de forma dosis dependiente la actividad de la lipoproteinlipasa, enzima que inactiva la lipólisis inducida por el glucagón y potencia el efecto lipogénico de la insulina, con lo que se favorece la formación de ácidos grasos, especialmente en el tejido adiposo omental79,80.
Experimentos con el ratón deficiente en el receptor GIP-R demostraron que tenía un fenotipo resistente a la obesidad inducida por la dieta, por lo que GIP puede estar involucrado en la patogenia de la llamada obesidad central. Así lo corroboran también estudios con ratones ob/ob en los que, además, se ha truncado el gen del receptor de GIP. En estos ratones se produce un menor incremento del peso y de la cantidad de tejido adiposo81,82. En el ratón knockout de GIP-R, también se ha descrito una menor incidencia de obesidad inducida por la extracción de los ovarios83.
Se está estudiando el empleo de antagonistas del receptor de GIP como método para reducir el peso corporal84,85. Sin embargo, hasta el momento los datos no son muy alentadores, pues un tratamiento crónico con estos antagonistas podría generar intolerancia a la glucosa o incluso hipertrigliceridemia.
GLP-1Es un pequeño péptido de 29 aminoácidos secretado por las células L del intestino, principalmente en íleon y colon (donde colocaliza con PYY y oxintomodulina) y también en zonas del sistema nervioso central. Se origina por procesamiento alternativo a partir de un precursor mayor, el preproglucagón, que es hidrolizado por las enzimas proconvertasas (PC1 y PC2), lo que origina distintos productos y fragmentos, según el tipo de tejido donde se lleve a cabo el proceso86, y con actividad biológica diversa.
La forma bioactiva mayoritaria en plasma es el GLP1(7–36)-amida. Se libera desde el intestino tras la ingestión de nutrientes, fundamentalmente hidratos de carbono y ácidos grasos y en proporción con el contenido calórico. Ejerce sus funciones a través de un único receptor descrito hasta la fecha86, el GLP-1R.
En roedores se ha observado que la administración de GLP-1 por vía central o periférica inhibe la ingestión de alimentos87 y un tratamiento prolongado causa pérdida de peso88. Estos efectos se pierden tras la administración de exendina (9–39), un antagonista del GLP-1R87. Sin embargo, el ratón deficiente en este receptor manifiesta una conducta alimentaria y un peso corporal normales, lo que se podría explicar por la redundancia en los mecanismos que controlan el apetito89,90. El GLP-1 también produce una reducción, dependiente de la dosis, del apetito y la ingestión de alimentos en humanos sanos, obesos o con diabetes mellitus91-93. La infusión intravenosa de GLP-1(7–36 )amida redujo en un 12% la ingesta de alimentos en humanos sanos no obesos y en pacientes diabéticos.
El GLP-1 también controla el balance energético a largo plazo. La administración subcutánea de GLP-1(7–36)-amida en humanos con diabetes mellitus redujo de forma significativa la ingesta y el peso corporal94. Esto mismo se observó en pacientes obesos, en los que el tratamiento con GLP-1 por vía subcutánea antes de cada comida durante 5 días redujo en un 15% la ingesta y produjo una pérdida de alrededor de 0,5kg de peso93.
La respuesta hipofágica inducida por el GLP-1 periférico se pierde tras la sección del nervio vago42,95. El GLP-1, a través de las fibras aferentes del nervio vago, envía la señal anorexigénica al núcleo del tracto solitario, desde donde parten proyecciones hacia neuronas del núcleo arcuato que estimulan la liberación de POMC y tránsito regulado por cocaína y anfetamina (CART), con lo que se inhibe la ingestión de alimentos96,97.
La reducción del apetito puede verse favorecida en parte por otras acciones que GLP-1 ejerce en el tracto gastrointestinal, donde inhibe la secreción de ácido gástrico inducida por la ingestión de alimentos, retrasa el vaciamiento gástrico y promueve la distensión gástrica, con lo que se crea sensación de saciedad86. Por otro lado, se ha comprobado que el GLP-1 puede causar aversión al alimento actuando en la amígdala del sistema límbico98,99.
En el páncreas, el GLP-1 inhibe la secreción de glucagón y estimula la biosíntesis de insulina incrementando su transcripción. El GLP-1 estimula además la síntesis de insulina dependiente de glucosa y es, conjuntamente con el GIP, una de las hormonas fundamentales en el efecto incretina100. Experimentos realizados en el knockout de GLP-1R reflejan una inadecuada respuesta insulínica a la administración oral de glucosa89.
Además, el GLP-1 regula la proliferación y la regeneración de las células beta en los islotes pancreáticos. Promueve la diferenciación de las células beta a partir de las células madre del epitelio del conducto pancreático. Además tiene efectos en el sistema cardiovascular incrementando la frecuencia cardíaca y la presión arterial, probablemente por activación del sistema nervioso simpático86.
La concentración de GLP-1 circulante es menor en individuos obesos, quienes presentan menos secreción posprandial del péptido que sus controles delgados. Cuando estos sujetos pierden peso, la concentración de GLP-1 se recupera101. Esta menor secreción de GLP-1 también se ha observado en mujeres adolescentes obesas y también en anoréxicas102.
Debido a su potente efecto insulinotrópico y a su actividad anorexigénica, el GLP-1ha suscitado gran interés como terapia en la diabetes mellitus tipo 2 y la obesidad. Sin embargo, uno de sus mayores problemas es su corta vida media en plasma (aproximadamente 2min), donde es transformado por la DPP IV en péptidos inactivos103. En busca de alternativas se han desarrollado análogos de la molécula de GLP-1 con mayor vida media100. Uno de esos compuestos es la liraglutida. Un tratamiento continuado con este análogo causó una pérdida de 1,2kg de peso y redujo la hemoglobina glucosilada un 2%, y se demostró su utilidad en el tratamiento de la diabetes mellitus tipo 2104,105. Otros análogos de GLP-1 como la exendina-4 han resultado eficaces en pacientes con diabetes mellitus tipo 2 que no consiguen un control glucémico adecuado con otros fármacos hipoglucemiantes. Exenatida es una molécula sintética modificada a partir de la exendina-4, péptido de 36 aminoácidos aislado del veneno del monstruo de Gila (Heloderma suspectum), que se une al GLP-1R y lo activa106. Es un agonista del GLP-1 resistente a la degradación por la DPP IV, lo que prolonga su vida media en plasma. Al igual que el péptido endógeno GLP-1, la exendina-4 es un potente secretagogo de insulina tanto en roedores como en humanos sanos o con diabetes mellitus tipo 2. En sujetos con diabetes tipo 2, el tratamiento con exenatida mejora el control glucémico y su administración por vía intravenosa tiene un efecto más duradero que el de GLP-1107. También comparte con el GLP-1 su capacidad anorexigénica. La administración intracerebroventricular de exendina-4 produce una inhibición, dependiente de la dosis, de la ingesta en varias especies108-110 y reduce el incremento de peso y la adiposidad en ratas Zucker tanto obesas como delgadas sin alterar la concentración de leptina111 y, además, es capaz de inhibir el incremento de ghrelina durante el ayuno en ratas Sprague-Dawley112. En pacientes con diabetes mellitus tipo 2, el tratamiento con exenatida durante 82 semanas consiguió una reducción de 4,4kg en el peso corporal113.
Oxintomodulina (OXM)Es un péptido de 37 aminoácidos que, al igual que GLP-1, se origina durante el procesamiento del gen del preproglucagón en las células L del intestino y en el sistema nervioso central114. Se aisló del intestino distal de cerdo, principalmente en yeyuno e íleon, y debe su nombre a su efecto inhibitorio en las glándulas oxínticas del estómago115.
Hasta la fecha no se ha descrito ningún receptor específico para OXM, pero dicho péptido parece ejercer sus efectos a través del receptor de GLP-1116,117. Así lo indican experimentos en los que se consigue bloquear algunas de las acciones de OXM (como su efecto anorexigénico) con exendina (9–39), antagonista del receptor de GLP-1118. Sin embargo, se ha comprobado que la afinidad de OXM por el GLP-1R es la mitad que la de GLP-1. A pesar de esto, el efecto inhibidor de GLP-1 y OXM sobre la ingesta de alimentos es similar, por lo que no se descarta la posible existencia de un nuevo receptor específico para OXM todavía no descrito117. Otro hecho que sostiene esta hipótesis es que GLP-1 y OXM parecen activar distintas regiones del sistema nervioso central, mientras OXM estimula neuronas situadas en el ARC, la acción de GLP-1 parece estar mediada en el tronco encefálico96. Sin embargo, existen estudios que demuestran la pérdida del efecto anorexigénico inducido por OXM en ratones deficientes en el receptor de GLP-190.
En las células L intestinales la OXM se expresa junto con otros péptidos anorexigénicos como PYY. Es liberada por el intestino al torrente sanguíneo tras la ingestión de alimentos, especialmente grasas, y su tasa de liberación es proporcional al número de calorías ingeridas. Su concentración se incrementa a los 30min del inicio de la comida y permanece elevada varias horas119,120.
En humanos la OXM tiene un potente efecto inhibitorio de la secreción ácida del estómago y el vaciamiento gástrico, y además produce distensión gástrica y genera sensación de saciedad. Por otra parte, inhibe la secreción exocrina pancreática e incrementa la secreción de insulina, proceso que requiere la integridad del nervio vago121,122.
La administración central o periférica de OXM en roedores tiene un potente efecto anorexigénico, por lo que se la considera una de las señales de saciedad que influyen a corto plazo en la ingestión de alimentos118,123. En este efecto anorexigénico a nivel central parece ser de gran importancia el PVN, pues cuando la OXM se administra directamente en esa región se requieren dosis más bajas que tras su administración intracerebroventricular. Se ha visto que la administración crónica de OXM durante 7 días reduce la ganancia de peso y la adiposidad y regula así la homeostasis energética a largo plazo124. La administración periférica de OXM produce la activación de neuronas en el ARC y la administración de exendina (9–39) directamente en el ARC bloquea ese efecto de la OXM123,125.
En humanos sanos, la infusión intravenosa de OXM antes de las comidas redujo en un 19% la ingesta calórica, reducción que persistió durante al menos 12h después de la administración126. Su infusión crónica redujo el peso corporal. Individuos obesos que recibieron inyecciones subcutáneas de OXM durante 4 semanas perdieron una media de 2,3kg de peso; por su parte, los sujetos que sólo recibieron placebo redujeron su peso unos 0,5kg127. Este efecto anorexigénico puede deberse en parte a la capacidad de OXM de reducir la ghrelina circulante, pues se ha observado que la administración de dosis posprandiales de OXM produce una reducción de un 15-20% en roedores y del 44% en humanos126. Por el contrario, la OXM no afecta a las concentraciones en ayuno o posprandiales de otras hormonas fundamentales en la regulación del balance energético, como la insulina, el glucagón, la leptina, el PYY y el GLP-1126.
La administración intravenosa de OXM reduce además la sensación de hambre durante el ayuno126. Los valores de OXM están elevados en condiciones asociadas a anorexia y pérdida de peso, como es el caso de la mala absorción tropical o tras la cirugía de bypass yeyuno-ileal128.
GLP-2También se origina a partir del precursor preproglucagón, al igual que el GLP-1 y la OXM. Se expresa fundamentalmente en el cerebro y se le han atribuido funciones tróficas y antiapoptóticas en la mucosa intestinal129,130. El GLP-2 circulante estimula la motilidad intestinal; sin embargo, sus efectos en el control de la ingesta no están claros. Algunos estudios han demostrado que la administración de GLP-2 a nivel central induce una respuesta hipofágica en ratas; no obstante, este efecto parece estar mediado por la activación del receptor de GLP-1, pues se desvanece tras la administración de un antagonista del GLP-1R131. Sin embargo, estudios posteriores no demostraron ningún efecto de GLP-2 en la ingesta o el peso corporal ni en roedores ni en humanos132,133, aunque sí se describió un descenso en la secreción de ghrelina tras la infusión intravenosa del péptido en humanos134.
HORMONAS OREXIGÉNICASGhrelinaA diferencia del resto de los péptidos descritos, la ghrelina es la primera hormona de origen gastrointestinal con capacidad activadora del apetito135. Este péptido de 28 aminoácidos se aisló en 1999 como ligando endógeno del receptor de secretagogos de GH (GHS-R)136.
La mayor parte de la ghrelina circulante procede de las células X/A-like de las glándulas oxínticas del fundus del estómago, aunque en menor medida se sintetiza también a lo largo del intestino, en ciertas regiones del sistema nervioso central o en tejidos periféricos como páncreas, riñones, corazón, placenta, sistema inmunitario, gónadas y pulmones137.
La molécula de ghrelina antes de ser secretada sufre una modificación postraduccional en el grupo hidroxilo de su serina de la posición 3, al que se añade un ácido n-octanoico. La ghrelina es la primera hormona conocida en mamíferos que sufre este proceso de acilación138. Esta modificación parece ser fundamental para algunas de las funciones biológicas de la ghrelina (entre ellas su efecto en la ingesta o la secreción de GH)139. Aunque inicialmente se creía que la ghrelina no acilada estaba desprovista de actividad biológica, se le han atribuido distintas funciones, como la estimulación de la adipogénesis y el control de la proliferación celular, y efectos en los sistemas cardiovascular y reproductor137.
La regulación de la secreción de ghrelina depende en gran medida del aporte de nutrientes. Durante el ayuno, los valores plasmáticos de ghrelina duplican los basales, para descender rápidamente tras la ingestión de alimentos y aumentar de nuevo progresivamente antes de la siguiente comida140,141.
Este perfil de secreción preprandial se mantiene incluso cuando se elimina el factor tiempo142. Así, en humanos, la ghrelina se incrementa antes del desayuno, la comida o la cena y se reduce inmediatamente después de cada comida140. Este ritmo en la secreción de ghrelina varía cuando se modifica el patrón de ingesta, lo que hace pensar que su secreción puede estar regulada por algún tipo de reflejo condicionado143.
La ghrelina, además de su incuestionable capacidad inductora de la secreción de GH, ejerce numerosas actividades biológicas en el organismo, en los ejes lactotropo, corticotropo y gonadotropo, modulando la actividad cardíaca, el ciclo sueño-vigilia y el metabolismo de lípidos y glúcidos. También se han demostrado sus efectos estimuladores de la motilidad gástrica y la secreción ácida, funciones que se bloquean tras vagotomía. Además, se le han atribuido efectos antiproliferativos y antiapoptóticos en diversos tejidos137.
La ghrelina es la señal orexigénica más potente descrita hasta la fecha. Su administración, ya sea central o por vía periférica, produce un incremento, dependiente de la dosis, de la ingesta en roedores144. Asimismo, la administración periférica de ghrelina genera una respuesta hiperfágica en humanos, en los que se incrementa en un 28% la energía consumida y aumenta la sensación de hambre145.
Pero la ghrelina también es importante en la regulación de la homeostasis energética a largo plazo. La administración periférica o intracerebroventricular crónica de ghrelina induce un incremento del peso corporal estimulando la deposición de grasas y, por lo tanto, incrementando la adiposidad en roedores146. In vitro se ha comprobado también que la ghrelina promueve la diferenciación de los preadipocitos y estimula la adipogénesis137.
En humanos, parece que la cantidad de ghrelina se correlaciona inversamente con la adiposidad. En sujetos obesos la concentración plasmática de ghrelina es menor que la observada en individuos normales147 y se restaura tras la pérdida de peso148. Sin embargo, los valores posprandiales de ghrelina no se reducen en sujetos obesos, lo que podría explicar la ingesta continua149. Por el contrario, en la anorexia nerviosa o en procesos de caquexia, las cifras de ghrelina son más altas que en individuos normales150,151. Tras la cirugía de bypass gástrico, los pacientes obesos presentan valores de ghrelina incluso más bajos, por lo que es probable que esta reducción de la ghrelina esté favoreciendo la disminución del apetito que se produce en estos sujetos148. Una excepción a la reducción de ghrelina en la obesidad es el síndrome de Prader-Willi, enfermedad que cursa con un fenotipo hiperfágico, obesidad mórbida, disfunción hipotalámica e hiperghrelinemia152. Este incremento desmesurado de la ghrelina podría explicar la hiperfagia característica de este trastorno153. Sin embargo, un estudio ha demostrado que, tras la administración de somatostatina en pacientes con Prader-Willi, se produce un descenso en los valores de ghrelina sin que se afecte la ingestión de alimentos154.
El efecto orexigénico de la ghrelina es independiente de su capacidad de estimular la secreción de GH y parece estar mediado por el hipotálamo. De hecho, la ablación del ARC con glutamato monosódico impide la ingestión de alimentos inducida por ghrelina155. La administración central o periférica de ghrelina incrementa en el ARC la expresión de NPY y AgRP (Agoutirelated protein), potentes estimuladores de la ingesta, e inhibe neuronas productoras de POMC y CART, neuropéptidos anorexigénicos144,156-158. El efecto orexigénico de la ghrelina también parece estar relacionado con las orexinas, pues su administración intracerebroventricular induce la expresión de c-fos en células inmunopositivas para orexinas, localizadas en el hipotálamo lateral159,160. El ratón deficiente en NPY y AgRP no responde a la administración exógena de ghrelina161.
A pesar de que el hipotálamo parece ser el centro principal que media la acción orexigénica de la ghrelina, hay indicios de que otras zonas extrahipotalámicas podrían participar en el proceso. Así, se ha detectado la expresión de GHS-R en neuronas del NTS y en el área postrema y se ha observado que la administración de ghrelina directamente en el cuarto ventrículo incrementa la ingesta de forma similar a cuando se administra en el tercer ventrículo. Además, se ha comprobado que su efecto orexigénico se pierde en pacientes que han sufrido una cirugía que conlleva vagotomía, por lo que el nervio vago parece ser un importante mediador de la acción de la ghrelina162-164.
Paradójicamente, el ratón deficiente en el gen de ghrelina y el que carece del receptor GHS-R1a no presentan alteraciones en el peso corporal, el tamaño y la conducta alimentaria, lo que se podría explicar por la existencia de sistemas compensadores165. Lo que sí se ha comprobado es que estos modelos de ratón evitan el desarrollo de una obesidad temprana cuando se exponen a una dieta rica en grasas, proceso que parece deberse a un incremento en el gasto energético166-168.
El empleo de antagonistas del receptor de ghrelina podría ser útil contra la obesidad. El tratamiento con el antagonista [D-Lys.3].GHRP-6 consigue inhibir la ingesta en un estado de ayuno. La administración crónica de este antagonista redujo el peso corporal del ratón ob/ob169. Por el contrario, el empleo de BIM-28163, antagonista del GHS-R1a, consiguió inhibir la liberación de GH, pero incrementando de forma paradójica el peso corporal. Otros estudios han empleado anticuerpos contra la ghrelina endógena y han conseguido retrasar la ganancia de peso en ratas170.
Del gen de la proghrelina deriva otro péptido denominado obestatina. Al igual que la ghrelina, la obestatina se ha aislado de estómago de rata171. Sin embargo, se han atribuido a la obestatina efectos contrarios a los de la ghrelina, de ahí su nombre. Se ha descrito que en roedores inhibe la ingestión de alimentos y el incremento de peso, además de retrasar el vaciamiento gástrico y disminuir la contractilidad intestinal172-174. Sin embargo, numerosos estudios no han conseguido reproducir estos resultados175-177. Se ha especulado que estas acciones de la obestatina podrían estar mediadas por el receptor huérfano GPR39178, aunque no todos los grupos concuerdan en esta afirmación. Por lo tanto, hay muchas discrepancias en los trabajos realizados con la obestatina, con lo que hasta que haya estudios más detallados es conveniente interpretar estos datos con cautela179-183.
CONCLUSIONESLas señales endocrinas originadas en el tracto gastrointestinal son fundamentales en la regulación de la ingestión de alimentos y la homeostasis energética. A excepción de la ghrelina, las hormonas gastrointestinales tienen efectos inhibidores del apetito. El conocimiento de los mecanismos fisiológicos y fisiopatológicos por los que estas moléculas regulan la ingestión de alimentos es fundamental para el desarrollo de nuevas estrategias en el tratamiento de los trastornos alimentarios. Sin embargo, que sean péptidos con una vida media muy corta en plasma y que se deba administrarlos por vía parenteral, junto con la existencia de mecanismos compensatorios que controlan el balance energético, hacen difícil su utilización a corto plazo. El desarrollo de análogos más estables en plasma, vías de administración más eficaces y un posible uso combinado podrían resultar útiles en el tratamiento de la obesidad.
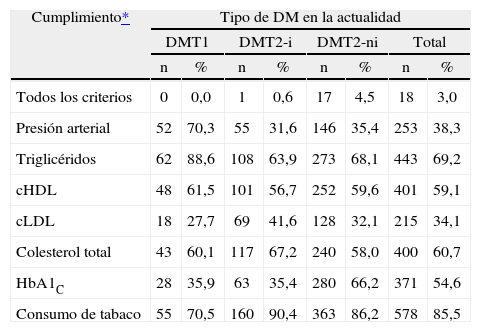
En el artículo original de Pedro de Pablos Velasco et al titulado "Estudio epidemiológico del perfil clínico y control glucémico del paciente diabético atendido en centros de atención primaria en España (estudio EPIDIAP)", publicado en Endocrinol Nutr. 2009;56(5):233–40, se han detectado los siguientes errores:
- −
Tanto en el resumen como en el abstract (p. 233), en el porcentaje que aparece en la última frase del apartado "Resultados", donde dice "5,4%" (resumen) y "5.4%" (abstract) debe decir "3%".
- −
En el texto:
- •
Página 236, apartado "Otras variables", primer párrafo, donde dice "5,4%" debe decir "3%".
- •
Página 239, segundo párrafo, primera línea, donde dice "5,8%" debe decir "3%".
- •
- −
En la tabla 5 (p. 238), se han detectado varios errores. A continuación se reproduce íntegramente la versión correcta de esta tabla:
Tabla 5.Cumplimiento de los criterios de la American Diabetes Association (ADA) de control para muestra total y según tipo de diabetes
Cumplimiento* Tipo de DM en la actualidad DMT1 DMT2-i DMT2-ni Total n % n % n % n % Todos los criterios 0 0,0 1 0,6 17 4,5 18 3,0 Presión arterial 52 70,3 55 31,6 146 35,4 253 38,3 Triglicéridos 62 88,6 108 63,9 273 68,1 443 69,2 cHDL 48 61,5 101 56,7 252 59,6 401 59,1 cLDL 18 27,7 69 41,6 128 32,1 215 34,1 Colesterol total 43 60,1 117 67,2 240 58,0 400 60,7 HbA1C 28 35,9 63 35,4 280 66,2 371 54,6 Consumo de tabaco 55 70,5 160 90,4 363 86,2 578 85,5 cHDL: colesterol de las lipoproteínas de alta densidad; cLDL: colesterol de las lipoproteínas de baja densidad; DMT1: diabetes mellitus tipo 1; DMT2-i: diabetes mellitus tipo 2 en tratamiento con insulina; DMT2-ni: diabetes mellitus tipo 2 no tratada con insulina; HbA1c: glucohemoglobina.
