


INTRODUCCIÓN
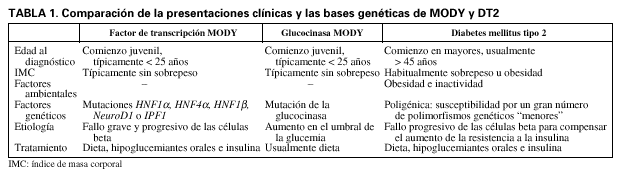
Maturity onset diabetes of the young (MODY) es un tipo de diabetes mellitus no insulino-dependiente causada por unas raras mutaciones autosómicas dominantes. Los pacientes MODY suelen presentarse antes de los 25 años y tienen típicamente unos nutridos antecedentes familiares de diabetes1. En la actualidad se han hallado mutaciones en 6 genes diferentes capaces de causar MODY: glucocinasa2-4, hepatocyte nuclear factor 1(HNF1α)5, HNF4α6, NHF1β7, insulin promoter factor 1 (IPF-1)8 y NeuroD19. Es probable que se describan otros genes, dado que se han hallado familias MODY sin mutaciones en ninguno de los genes anteriores. Las mutaciones en los genes diferentes tienen un curso clínico variable y rasgos extrapancreáticos y responden al tratamiento medicamentoso10,11. La diabetes tipo 2 (DT2) difiere de los 2 subgrupos principales de MODY (tabla 1). Es posible que los pacientes MODY se diagnostiquen erróneamente de DT2, especialmente si el diagnóstico es tardío y/o son obesos; sin embargo, es un diagnóstico erróneo, pues los pacientes con una base monogénica definida no pueden presentar DT2, tal y como se define ésta en la actualidad. Aunque MODY y DT2 son 2 trastornos independientes, el conocimiento de MODY puede ayudar a comprender los mecanismos patogénicos o la genética molecular de la DT2.

La diabetes tipo 2 es una enfermedad multifactorial. Los estudios efectuados en gemelos, en familias y en emigrantes sugieren que las diferencias genéticas pueden explicar una considerable proporción de las variaciones interindividuales en la susceptibilidad a la DT212. A diferencia de MODY, donde una sola mutación puede causar la enfermedad, el componente genético de la predisposición a la DT2 está compuesto por múltiples variantes de susceptibilidad, cada una de ellas con un pequeño efecto sobre la predisposición a la enfermedad. Las interacciones gengen y genambiente son asimismo importantes. Aunque la naturaleza poligénica y heterogénea de la DT2 complica la identificación de los genes que intervienen en la susceptibilidad a la DT2, el logro de dicha identificación mejorará nuestros conocimientos acerca de la fisiopatología subyacente. Ello serviría de ayuda para mejorar el tratamiento, como ya está empezando a ocurrir con MODY11.
Mediante la clonación posicional se ha logrado identificar las mutaciones que intervienen en muchas enfermedades monogénicas13. Con la clonación posicional en familias numerosas, se observó que MODY3 se producía por mutaciones en el gen HNF1α5; y MODY1, por mutaciones en el gen HNF4α6. En las enfermedades monogénicas como MODY, causadas por mutaciones únicas y en las que se dispone de familias numerosas, la clonación posicional constituye un método valioso. En la DT2 se han llevado a cabo diversas investigaciones amplias del genoma. A diferencia de la diabetes tipo 1, en la que se ha identificado la región HLA como el principal locus predisponente14, no se han demostrado grandes picos únicos de vinculación, y el progreso en la clonación posicional de los genes de la DT2 ha sido lento. En la actualidad, el gen Calpain-10 es el único gen de la DT2 que se ha descubierto mediante una investigación global del genoma15,16. En las investigaciones del genoma, la replicación uniforme de señales de vinculación relativamente pequeñas implica una variación en diversas regiones cromosómicas17, pero el éxito limitado de la clonación posicional sugiere que los estudios sobre los genes candidatos son también importantes para identificar las variables de susceptibilidad a la DT2.

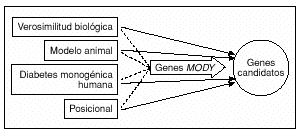
Fig. 1. Los genes MODY cumplen todos los requisitos de un buen gen candidato para la DT2.
Son diversos los motivos por los cuales puede considerarse a un gen como candidato a la susceptibilidad para la DT2 (fig. 1), a saber: el conocimiento del papel biológico de las proteínas codificadas; puede constituir un candidato posicional si se encuentra bajo un pico de vinculación; puede quedar implicado en los estudios de animales transgénicos; o bien, porque las mutaciones graves dan lugar a la diabetes monogénica humana. Los genes MODY pueden quedar incluidos en todas estas categorías. Como hecho importante, dado que las mutaciones heterocigóticas en los genes MODY dan lugar a diabetes, ello demuestra que la proteína que codifican es un componente bioquímico clave de la función normal de las células beta, y que la disminución de su valor no puede compensarse. Las variantes comunes de los genes MODY son unos firmes candidatos a desempeñar un papel en la DT2 humana y en los rasgos afines a la misma, debido a que un pequeño cambio en la expresión de uno de estos genes, o una alteración en la actividad de la proteína expresada, predisponen probablemente a la elevación de la glucemia. En esta revisión se consideran los estudios recientes sobre las variaciones comunes de los genes MODY, como método para identificar las variantes de susceptibilidad genética a la DT2.
GENES MODY EN LA DIABETES TIPO 2
Cinco de los genes MODY son factores de transcripción de las células beta; el sexto es una enzima glucolítica, la glucocinasa. En la tabla 2 se ofrecen descripciones de los 6 genes MODY, junto con un resumen de las variantes comunes de estos genes que se han asociado convincentemente con la DT2 o con un rasgo afín.
Fig. 2. El alelo S particular en G319S predispone a la diabetes del tipo 2 (DT2) en la población Oji-Cree. El alelo S no es una mutación MODY, dado que: (a) se halla presente en la población Oji-Cree normal e incrementa el riesgo de la DT2 en un riesgo relativo de 2 aproximadamente, y (b) el polimorfismo causa sólo una moderada disminución en la eficacia de la transactivación26. IMC: índice de masa corporal.
GENES MODY DEL FACTOR DE TRANSCRIPCIÓN
Los genes MODY del factor de transcripción de las células beta forman parte de una red reguladora que es esencial para el desarrollo, la diferenciación y la supervivencia adecuados de las células beta18. En contraste con las mutaciones de la glucocinasa, las mutaciones de HNF1α, HNF1β, HNF4α, IPF1 y NeuroD1 causan una grave disfunción progresiva de las células beta1 (tabla 1).
HNF1α
Las mutaciones en HNF1α son la causa más frecuente de MODY en el Reino Unido, siendo responsables del 63% de los casos aproximadamente19. Existen pruebas convincentes de que las variantes comunes del gen HNF1α son también importantes para la susceptibilidad a la DT2. En un amplio estudio del genoma de familias finlandesas DT2 insulino-deficientes se identificó el pico de vinculación NIDDM2, que coincide con el locus MODY3 en el cromosoma 12q2420. En algunas investigaciones posteriores efectuadas en poblaciones diferentes también se han hallado pruebas de vinculación en esta región genómica17,21.
Variación común en HNF1α y DT2 en la población Oji-Cree
La prueba más convincente del papel que desempeñan las variantes comunes de HNF1α en la predisposición a la DT2 proviene de la población Oji-Cree de North-Western Ontario y Manitoba, Canadá. Esta población india nativa, que vive aislada, tiene una de las mayores prevalencias mundiales de diabetes tipo 222. Se ha visto que una variante del gen HNF1α, G319S, que es particular del grupo Oji-Cree y común en el mismo, predispone en gran medida a la diabetes tipo 223. La frecuencia del alelo S319 es aproximadamente del 9% en los controles no diabéticos, y del 21% en los diabéticos de tipo 2 (p < 0,001) (fig. 2a). La probabilidad relativa de DT2 en los heterocigóticos G319S es de 1,97 (1,44-2,70), y de 4,00 (2,65-6,03) en los portadores homocigóticos de S319, en comparación con los homocigóticos del tipo natural. En las mutaciones MODY3, la haploinsuficiencia es el mecanismo más habitual de la mutación25. Suele haber una grave pérdida funcional (> 95%) del alelo mutado. En contraste con las mutaciones MODY3, los estudios funcionales in vitro han demostrado que el produc to alélico S319 HNF1α da lugar a una reducción aproximada del 50% en la actividad de transactivación26 (fig. 2b). Dado que esta variante sólo causa una pérdida parcial en la función de HNF1α, para que se desarrolle la diabe tes es necesaria la presencia de otros factores de estrés, como la obesidad y la resistencia a la insulina; ello explica que los pacientes MODY3 y los diabéticos tipo 2 de la población Oji-Cree tengan unos fenotipos clínicos diferentes26.
Fig. 3. Representación esquemática del gen HNF4α, con el promotor P2 alternativo. El isómero predominante en el hepatocito es HNF4α1, que utiliza el promotor P1 y el exón 1a. En contraste, el isómero predominante en la célula beta es HNF4α, que utiliza el promotor P2 y el exón 1 alternativo. En ambos isómeros quedan escindidos los exones 1c y 1b.
Variantes comunes de HNF1α y DT2 en otras razas
Aparte de la población Oji-Cree, no se ha identificado ninguna otra variante comparable de HNF1α en la susceptibilidad a la DT2. En la población de raza blanca existen 3 polimorfismos sin sentido comunes: A98V, I27L y S487N27. La variante A98V se asoció con una disminución del 20% en la respuesta de los valores plasmáticos del péptido C y de la insulina a los 30 min en la prueba de tolerancia a la glucosa oral (PTGO), realizada en 240 individuos daneses de raza blanca y de mediana edad con tolerancia normal a la glucosa (TNG)28. En otra cohorte de 377 sujetos jóvenes con TNG, esta asociación se observó solamente en individuos homocigóticos V98 (de los cuales sólo había 2, pero que demostraron unas disminuciones del 39 y del 44% en la respuesta aguda del péptido C en la PTGO). El estudio no aportó pruebas sobre un papel predisponente a la DT2 (245 casos frente a 240 controles, probabilidad relativa (PR) = 0,83 [0,44-1,58], p = 0,69). El resultado del péptido C a los 30 min se reprodujo en un segundo estudio de 231 sujetos TNG, familiares de primer grado de probandos con DT229. En cambio, en un estudio de 156 individuos chinos y 222 finlandeses30 se demostró una asociación significativa del alelo V con la DT2 (PR en los sujetos chinos = 2,69 [0,32-∞; PR en los sujetos finlandeses = 2,20 [0,93-5,20], p combinada = 0,01), pero en 295 sujetos TNG y en 38 con intolerancia a la glucosa (IG) no se demostró que disminuyera la función de las células beta (aunque los autores no presentaron los datos sobre el péptido C). La reproducción del resultado de A98V se ha visto dificultada por su baja frecuencia (frecuencia del alelo menor [FAM] ~4% en los europeos de raza blanca) y por su aparente ausencia en los individuos chinos31, judíos ashkenazi32 e indios pima33. Es necesario un estudio a gran escala para confirmar la asociación con la función de las células beta y la DT2. No hay pruebas de que la variante S487N (FAM 30-45%) influya sobre la función de las células beta o sobre el riesgo de presentar DT227,28,30,32-35).
Se ha observado una asociación variable del polimorfismo I27L con la diabetes tipo 2 (FAM 25-45%)27,28,30,32-35. No se halló ninguna diferencia significativa en la frecuencia de la variante en un estudio inicial de 245 pacientes con diabetes mellitus no insulino-dependiente (DMNID) y en 250 sujetos de control, de edad similar, con tolerancia a la glucosa (PR = 1,29 [0,98, 1,70], p = 0,09)27. En estudios realizados posteriormente en poblaciones de individuos chinos (PR = 1,55 [0,96-2,51], p = 0,09)30, finlandeses (PR = 1,27 [0,86-1,87], p = 0,27)30, judíos ashkenazi (PR = 1,09 [0,75-1,57], p = 0,76)32 e indios pima (PR = 0,91 [0,54-1,53]33 tampoco se observó asociación, pero en todos los casos se trataba de grupos pequeños. Aunque estos estudios no han aportado individualmente diferencias significativas, en conjunto la literatura publicada apoya al parecer un ligero efecto del alelo Leucine sobre la predisposición a la DT2 (PR ~ 1,2). Dado que todos los estudios realizados hasta el momento no han tenido la potencia suficiente para detectar una PR de dicha cuantía, es necesaria la confirmación con un metaanálisis y con un estudio de replicación cuya potencia sea suficiente.
Hay algunas pruebas de que la variante I27L influye sobre la función de las células beta. En un estudio realizado en individuos daneses que eran familiares en primer grado de probandos con DT2, en la PTGO se halló a los 30 min una disminución del 32% en la concentración sérica del péptido C (p = 0,01), y un descenso del 39% la concentración sérica de insulina (p = 0,02)34. Sin embargo, en una cohorte 3 veces mayor, de 230 sujetos con TNG, hijos de probandos que presentaban DT2, no se reprodujeron estos resultados34. En un estudio posterior realizado en 60 sujetos de raza blanca con tolerancia a la glucosa se halló un efecto significativo del polimorfismo I27L sobre la primera y la segunda fase de la respuesta insulínica35, aunque en otros estudios a mayor escala no se halló asociación con la función de las células beta28,30.
Cada vez se pone más de manifiesto que es necesario realizar estudios de asociación bien diseñados, con potencia suficiente y a gran escala para detectar la reproducibilidad de las pequeñas señales poligénicas36-38, y se espera la confirmación o la refutación, a una escala suficiente, del efecto de estas variantes HNF1α. Incluso aunque A98V y/o I27L sean variantes causales y desempeñen algún papel en la patología molecular de la DT2, probablemente no explican la señal de vinculación observada en el locus HNF1α; es probable que tengan importancia a este respecto otras variantes HNF1α, mudas o no codificantes. Al igual que ocurre con todos los genes candidatos, las respuestas definitivas sobre el papel de la variante común HNF1α en la DT2 sólo se lograrán mediante un análisis detallado del desequilibrio de ligamiento y de la estructura de los haplotipos de la región, seguido de estudios de asociación a gran escala basados en el marcaje de los haplotipos SNP (htSNP)39.
HNF4α
Varios análisis amplios del genoma en la DT2 han demostrado la existencia de ligamiento a 20q12-13.117, donde reside el gen HNF4α. En el análisis inicial de las regiones de codificación y del promotor P1 no se identificó ninguna variante de HNF4α que se asociara significativamente con la DT2 y explicara la ligamiento observada40,41. Sin embargo, el descubrimiento y la caracterización de un promotor alternativo, P2, ha llevado a reanalizar el papel del gen HNF4α en la DT242. La figura 3 muestra un diagrama esquemático del gen HNF4α, con los promotores P1 y P2. El promotor P2 ocurre a 46kb de distancia, en sentido anterógrado, del sitio inicial de transcripción de HNF4α, y se ha observado que es activo en las células beta pancreáticas y en los hepatocitos42. Las transcripciones que surgen del promotor P1 son más abundantes en los hepatocitos; en cambio, en las células beta predomina la variante de escisión P2 HNF4α. El promotor P2 contiene sitios de ligamientos funcionales para los productos de los genes MODY del otro factor de transcripción, y se ha observado que las mutaciones en los sitios IPF-1 y HNF4α en el promotor P2 se co-segregan con MODY42,43. Por lo tanto, el promotor P2 es un importante nodo en la compleja red reguladora de las células beta que conecta los factores de transcripción MODY. Se ha sugerido que esta alternancia 5' final de la unidad de transcripción HNF4α podría servir de gran objetivo para la interferencia transcripcional, y que las variantes en esta región podrían explicar el ligamiento de la diabetes tipo 2 con esta área genómica42.
En un análisis inicial del promotor P2 y del primer exón alternativo, en sujetos con diabetes tipo 2 de comienzo juvenil y en probandos de familias con ligamiento al cromosoma 20, se identificaron sólo variantes raras que carecían de significado biológico44. Sin embargo, en un estudio reciente efectuado en una población de judíos ashkenazi en la que se había demostrado previamente ligamiento al cromosoma 20q12-13.1 y donde se utilizó el marcaje del haplotipo SNP de una región de 78kb en torno a <HNF4α, se halló una asociación de 2 de los htSNP con la DT245. Uno de ellos era un SNP intrónico 3' (frecuencia del alelo menor 29,2% en los casos, frente a 21,7% en los controles, p = 0,0028, PR 1,49 [1,15-1,90]). Como hecho de sumo interés, el segundo SNP (P2 SNP) se localizó aproximadamente a 3,9 kb de distancia, en sentido anterógrado, de P2 (frecuencia del alelo menor 26,9% en los casos, frente a 20,3% en los controles, p = 0,0078, PR 1,46 [1,12-1,91]). Se observó que P2 SNP formaba parte de un bloque haplotípico > 10kb asociado a la DT2, que se extendía a 5' del promotor P2. En las familias donde el probando es portador al menos de un alelo de riesgo, P2 SNP puede ser responsable de la mayor parte de la señal de ligamiento observado en el cromosoma 20q en esta población. Es importante señalar que la asociación P2 SNP y el perfil de ligamiento fraccionado se observaron independientemente en una muestra finlandesa46, lo que habla muy a favor de la presencia de variantes del elemento regulador que contribuyen al riesgo de la DT2. Como la región del promotor P2 parece ser un nodo clave en la red de células beta pancreáticas, es posible que ocurran interacciones gen-gen con variantes en los otros genes MODY.
Este trabajo sobre HNF4α demuestra asimismo la importancia de efectuar un análisis global del desequilibrio de ligamiento y del haplotipo en la región de un gen candidato. La funcionalidad de la región anterógrada del promotor P2 no era evidente, y pasó por alto la asociación P2 SNP al concentrar la atención sobre los "SNP candidatos" en la región del codificación del gen y en las inmediaciones de la misma.
IPF-1
IPF-1 (conocido también como PDX1) es clave en el desarrollo del páncreas y también en los islotes maduros, donde interviene en la transcripción del gen de la insulina47,48. La importancia de IPF-1 en la función pancreática humana queda demostrada por el efecto de las mutaciones raras: los portadores de mutaciones homocigóticas se presentan con agenesia pancreática49, y los mutantes heterocigóticos desarrollan MODY8. Se ha observado que una mutación en el sitio de unión IPF-1 del promotor P2 HNF4α cosegrega con MODY42. Un estudio inicial de asociación sugirió que 3 polimorfismos de codificación, C18R, D76N y R197H, se presentan aproximadamente en el 1% de la población de raza blanca del Reino Unido, y que los sujetos predispuestos a la diabetes tipo 2 tienen un riesgo relativo de 350. En trabajos in vitro se sugirió además que estas variantes influyen sobre la función de IPF-150. En estudios realizados en Francia y Suecia se mostró también que las mutaciones funcionales predisponían a la diabetes tipo 2, especialmente cuando los sujetos se diagnosticaban antes de los 50 años de edad51,52. La baja frecuencia (en algunas poblaciones estas variantes se hallaban ausentes) y las múltiples variantes detectadas indican que los estudios tienen una escasa potencia estadística; ello puede explicar que en otros estudios no se hayan reproducido estos hallazgos53-55.
NEUROD1 Y HNF1β
Se han realizado escasos análisis sobre el papel de NeuroD1 y HNF1β en la DT2. En las poblaciones cribadas hasta el momento, la única variante común (FAM > 5%) de codificación NeuroD1 que se ha identificado es Ala45Thr. NeuroD1 ocurre en las proximidades del pico de ligamiento IDDM7, y la mayoría de los trabajos se han enfocado sobre el papel de esta variante en la diabetes tipo 1. Como ocurre típicamente en los estudios de asociación genética38, el estudio positivo inicial sugirió un papel importante del alelo Thr45 en la predisposición a la diabetes tipo 1 (DT1)56, pero el apoyo a esta hipótesis fue generalmente escaso o nulo en estudios posteriores57-61. En conjunto, la literatura publicada apoyaba al parecer la existencia de una asociación, hasta que en un estudio a gran escala se observó que ello era improbable61a. La prueba de que las variantes de NeuroD1 desempeñen un papel en la susceptibilidad a la DT2 es más limitada59,62, pero se está a la espera de un análisis global de este gen. Lo mismo cabe decir de HNF1β, sobre el cual se han realizado pocos estudios63,64.
FACTOR MODY NO DE TRANSCRIPCION: GLUCOCINASA
Las mutaciones en el gen de la glucocinasa (GCK) causan MODY21. GCK codifica a la glucocinasa que, en las células beta pancreáticas y en los hepatocitos, cataliza el primer paso limitante del metabolismo de la glucosa. El papel regulador clave de la glucocinasa en las células beta ha conducido a que se describa como el "sensor de la glucosa en las células beta pancreáticas"65. El fenotipo MODY2 se caracteriza por una leve hiperglucemia en ayunas (usualmente entre 5,5 mmol/l-8,5 mmol/l) que dura toda la vida, aunque empeora escasamente con la edad1. A menudo los pacientes no requieren tratamiento y la concentración de glucosa se encuentra habitualmente tan sólo justo por encima del rango normal de la población. La fisiopatología subyacente consiste en un aumento del umbral de la glucosa plasmática, debido a una disminución en la sensibilidad de la glucocinasa frente a la glucosa. La secreción de insulina queda regulada de acuerdo con este umbral más alto de glucemia66. Además de causar una ligera hiperglucemia, las mutaciones de la glucocinasa afectan también al peso de nacimiento67. Los hijos de madres con una mutación de la glucocinasa presentan un mayor peso al nacer (aumento ~ 600 g), debido a un aumento de la secreción fetal de insulina en respuesta a la hiperglucemia materna. A la inversa, si el feto hereda una mutación GCK, disminuye su sensibilidad a la glucosa, se reduce la secreción de insulina fetal y estos niños pesan por término medio 500 g menos al nacer.
El efecto de las mutaciones raras sobre la sensibilidad a la glucosa y sobre el peso de nacimiento sugiere que las variantes comunes de GCK pueden explicar algunas de las variaciones de estos rasgos cuantitativos dentro de una población. Al secuenciar a 100 individuos de raza blanca del Reino Unido no emparentados, no se observaron polimorfismos comunes (FAM > 5%) en la región de codificación de GCK (Weedon, Ellard y Frayling, datos no publicados). Por lo tanto, la mayoría de los estudios de asociación de GCK se han concentrado en una variante A a G, con frecuencia del alelo menor del 18%, en la región del promotor de las células insulares, GCK(-30). GCK(-30) ocurre a 30bp de distancia de GCK, en sentido anterógrado, en una región donde se halla una estrecha homología entre el ser humano, el ratón y la rata (sitio web Santa Cruz: http://genome. ucsc.edu) y se ha observado que la mutagénesis de transversión de una secuencia de 10bp que incluye a GCK(-30) afecta a la expresión de GCK68. Por lo tanto, es probable que GCK(-30) ejerza una cierta influencia sobre la expresión de GCK. Se han realizado diversos estudios de asociación sobre la variante GCK(-30)69-76, pero los resultados han sido contradictorios. Estos estudios se han concentrado sobre la asociación con mediciones dinámicas de la tolerancia a la glucosa, la intolerancia a la glucosa y la DT2. Las pruebas realizadas sobre rasgos cuantitativos no tan directamente relacionados con el fenotipo monogénico, junto a su tamaño relativamente pequeño, explican que estos estudios previos de GCK(-30) no hayan mostrado asociación. En 4 de 8 estudios publicados se han presentado resultados de glucosa plasmática en ayunas (GB). En el estudio de menor tamaño70 (n = 65) se demostró una asociación significativa del alelo A con la cifra de GB (GG = 5,3 mmol/l, frenta a A* = 5,8 mmol/l, p < 0,05) en sujetos con TNG en situación basal; sin embargo, los resultados no se reprodujeron en un seguimiento de 5 años. En los otros 3 estudios69,72,75 no se observaron tendencias significativas de aumento de la GB con el alelo A en GCK(-30).
Más recientemente, mediante grandes cohortes de individuos normoglucémicos (n total = 2.000), hemos demostrado que GCK(-30) contribuye verdaderamente a la variación poblacional en los valores de glucemia en ayunas. Los portadores de un alelo A en GCK(-30) tienen unas cifras de glucosa en ayunas (GB) aproximadamente 0,08 mM más elevadas que los sujetos homocigóticos para el alelo G77. Además, al estudiar a 5.567 madres e hijos hemos demostrado que las madres portadoras de un alelo A en GCK(-30) tienen hijos que pesan 68 g más al nacer77. Dado el efecto relativamente pequeño de la variación GCK(-30) sobre la GB, es improbable que ejerza una influencia importante sobre la susceptibilidad a la DT2. Sin embargo, ello constituye un ejemplo excelente del modo como los alelos comunes de un gen, en el que las mutaciones raras causan un trastorno monogénico, pueden ejercer unos pequeños efectos, aunque prevalentes, sobre un rasgo poligénico relacionado.
Es posible que GCK(-30) no sea la única variante de GCK que influye sobre la actividad de la glucocinasa: otros polimorfismos GCK pueden contribuir a la variación poblacional de los valores de glucemia y del peso de nacimiento. Es necesario realizar estudios en grandes cohortes sobre haplotipos y desequilibrios de ligamiento para identificar los SNP más informativos en la región GCK, y tipificar estos haplotipos que marcan los SNP (htSNP).
OTROS GENES "MONOGÉNICOS" Y DIABETES TIPO 2
Además de MODY, se ha observado que variantes comunes de otros genes donde las mutaciones raras causan un fenotipo relacionado con la diabetes, predisponen a la DT2. Las mutaciones en PPAR( causan una grave resistencia a la insulina78, y la variante común Pro12Ala fue el primer polimorfismo que se asoció consistentemente con la DT2; el alelo Pro12 confiere una probabilidad relativa de 1,2536. Se ha observado que las mutaciones con pérdida de función en el gen Kir6.2 causan el hiperinsulinismo familiar79,80, y recientemente se ha observado también que las mutaciones activantes de Kir6.2 son una causa común de la diabetes neonatal permanente de los jóvenes (DNPJ)81. Existen pruebas convincentes de que el alelo K del polimorfismo co mún E23K predispone a la diabetes tipo 2 con una PR de ~1,237,82.
CONCLUSIÓN
El hecho deas mutaciones heterocigóticas de los genes MODY den lugar a diabetes demuestra que la pérdida parcial de función de este gen no puede compensarse por otras vías bioquímicas o celulares en las células beta pancreáticas. Por lo tanto, las variantes comunes de los genes MODY se hallan entre los candidatos más idóneos para explicar una parte de la variación poblacional en el riesgo para la DT2 o para un rasgo poligénico afín. En la presente revisión se ha ilustrado lo dicho, al mostrar que las variaciones comunes de MODY y de otros genes de la diabetes monogénica predisponen a la DT2 e influyen sobre los rasgos afines a la diabetes. Además, es necesario realizar análisis globales a gran escala sobre el papel de los genes MODY en la susceptibilidad a la DT2.
AGRADECIMIENTOS
Deseamos dar las gracias a nuestros colegas en Exeter y en los otros centros que han contribuido a nuestros estudios sobre MODY y la genética de la diabetes tipo 2. También agradecemos el gentil apoyo económico recibido de Diabetes UK y Wellcome Trust para el trabajo genético realizado en Exeter, descrito en este estudio. ATH es un Wellcome Trust Clinical Research Leave Fellow.
