






El seudohipoparatiroidismo (PHP) comprende un grupo heterogéneo de enfermedades endocrinas que se caracterizan por hipocalcemia, hiperfosfatemia y resistencia a la paratirina (PTH)1. Hay diferentes tipos de seudohipoparatiroidismo; en algunos de ellos se ha aclarado el trastorno molecular subyacente permitiendo un correcto diagnóstico y un adecuado consejo genético. Con el fin de entender los diferentes tipos de seudohipoparatiroidismo conviene recordar que el mecanismo de acción de la PTH se basa en su unión a un receptor de membrana que está acoplado a una proteína G (fig. 1). La proteína G estimuladora (Gs) es miembro de la familia de proteínas G, y está constituida por la subunidad alfa específica que se une al nucleótido guanina del grupo GTP/GDP e interacciona con los receptores y efectores específicos y las subunidades beta y gamma que forman el complejo necesario para la activación de Gsµpor los receptores2,3. La función de la proteína Gs es transmitir señales desde los receptores de la superficie celular hasta los efectores intracelulares (la adenilato ciclasa en el caso de la PTH) que generan segundos mensajeros (AMPc). La causa genética de varios de los diferentes tipos de PHP, definidos con criterios clínicos, es un trastorno en la proteína Gsµ.
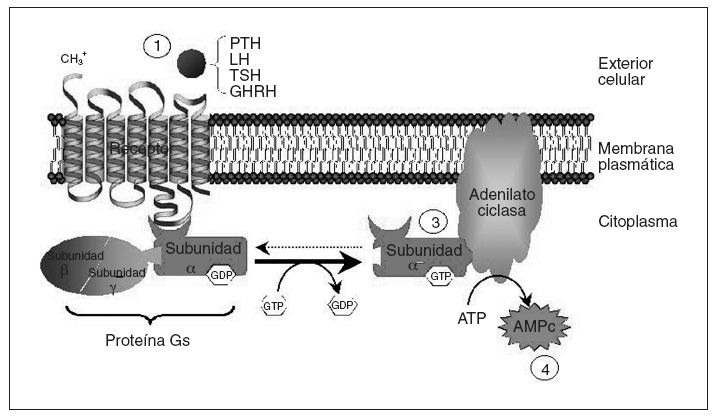
Fig. 1. Esquema del mecanismo de acción de hormonas mediado por receptores acoplados a proteína Gs. La unión de la hormona a su receptor (1) causa la activación de la subunidad alfa de la proteína Gs (2), que interaccionará con la adenilato ciclasa (3), produciendo la síntesis de AMPc (4), que funcionará como segundo mensajero transmitiendo la información procedente del estímulo hormonal. Tras la activación de la adenilato ciclasa, la subunidad alfa de la proteína Gs vuelve a su estado basal. AMPc: adenosinmonofosfato cíclico; ATP: adenosintrifosfato; GHRH: somatoliberina; LH: Lutropina; TSH: tirotropina.
Por otra parte, además de la PTH, otras muchas hormonas usan receptores acoplados a la proteína Gsµy la adenilato ciclasa para generar AMPc, por lo que una alteración puede causar no solamente resistencia a PTH (y consecuentemente seudohipoparatiroidismo), sino también a tirotropina (TSH), lutropina (LH), folitropina (FSH), somatoliberina (GHRH), vasopresina (ADH), glucagón, corticotropina (ACTH) y calcitonina, entre otras, con el correspondiente fenotipo asociado en cada caso4,5. A continuación revisaremos la base molecular y los trastornos genéticos relacionados con el PHP.
Estructura del gen GNASEl gen que codifica para la proteína Gsµes el gen GNAS, situado en el locus GNAS, en el brazo largo del cromosoma 20 (20q13.2-13.3)6. Estructuralmente el locus GNAS está formado por 12 exones comunes (exón 2 a exón 13 de la proteína Gsµ) que se unen a uno de los primeros cuatro exones alternativos. En función de qué primer exón alternativo se una a los restantes exones (2-13) y se transcriba, se producirán cuatro tipos diferentes de proteínas o de transcritos (fig. 2):
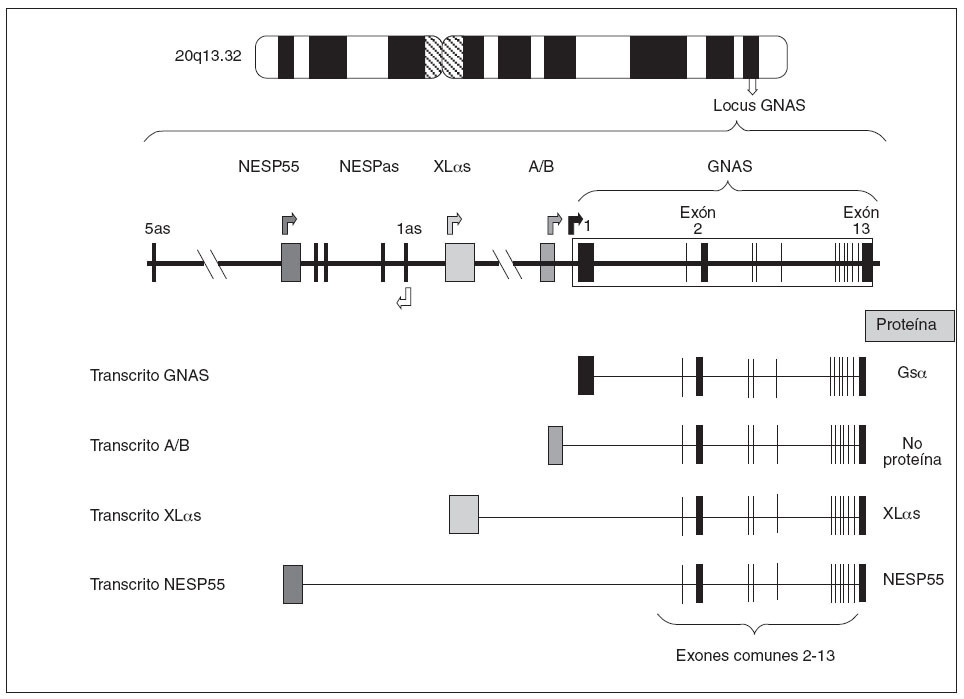
Fig. 2. Organización del locus GNAS. Las cajas y las líneas verticales representan los exones. Entre los exones (línea horizontal) están los intrones. Las flechas indican el punto de comienzo de lectura de los cuatro transcritos principales. Como puede observarse, los cuatro transcritos sólo difieren en el primer exón 1 alternativo, mientras que los exones 2-13 son comunes (el dibujo es orientativo, no hemos de olvidar que en el procesado del ARNm se eliminan los intrones). El exón 1, al unirse a los exones comunes 2-13, codifica para la propia proteína Gs α; el exón A/B, al unirse al resto de los exones comunes, genera un transcrito que no codifica para ninguna proteína; el exón XL αs, al unirse a los exones 2-13, codificará para la forma larga de la proteína Gs α(XL αs), y el exón codificante de NESP55.
– Así, el primer exón más cercano al resto del gen es el llamado exón 1 de Gsµque, al unirse a los exones restantes comunes (2-13), forma la propia proteína Gsµ. – Una segunda posibilidad es que otro exón 1 alternativo, también llamado exón A/B o exón 1A, situado 2,5 kb antes del exón 1 de Gsµ, se empalme con el resto de los exones comunes 2-13. En ese caso, debido a que no hay un comienzo de traducción AUG en el exón A/B, se piensa que el transcrito resultante no es traducido7,8. – Una tercera posibilidad es que otro exón 1 alternativo, situado a 35 kb del exón 1 de Gsµ, se una a los exones 2-13 dando un transcrito que codifica la proteína XLµs, una isoforma de Gsµcon similares funciones pero ligeramente más larga9,10. – Por último,está el exón alternativo más alejado (a 49 kb del exón 1) que, unido al resto de los exones comunes 2-13, produce el transcrito codificante para la proteína NESP55, proteína similar a la cromogranina que se expresa en los tejidos neuroendocrinos9.
Imprintingdel locus GNASEl locus GNAS y los diferentes transcritos para los que codifican (proteínas Gsµ,XLµs, NESP 55 y transcrito A/B) están sometidos a imprinting o impronta genética, por lo que el patrón de herencia del seudohipoparatiroidismo cuando se produce por trastornos de la proteína Gsµ, como veremos a continuación, estará regido por este fenómeno de imprinting.
Generalmente, la mayoría de los genes funcionan a través de la expresión de sus dos alelos (materno y paterno). Sin embargo, algunos genes tienen uno de sus dos alelos inactivado o improntado o con fenómeno de imprinting (o sea, el individuo hereda los dos alelos, pero uno de ellos no se expresa o está inactivo). Si se trata del alelo heredado de la madre, hablamos de que ese gen sufre imprinting materno (en este caso, el individuo es normal solamente con la expresión genética del alelo paterno, ya que el alelo materno está inactivo), y viceversa, si el inactivo es el heredado del padre, hablaríamos de imprinting paterno (en este caso, el individuo es normal solamente con la expresión genética del alelo materno).
Esta expresión genética diferencial puede durar toda la vida o estar limitada a un estadio del desarrollo, y puede ser generalizado para todos los tejidos donde se expresa el gen o puede ser tejido dependiente (o sea, un gen puede mostrar su carácter improntado sólo en determinadas células, o en un tiempo concreto del de-sarrollo)11,12. En la mayoría de los casos el efecto imprinting (o inactivación de un alelo) se debe a que ese alelo está metilado (adición de grupos metilo en citosinas); en otras ocasiones el mecanismo de imprinting es desconocido.
Como indicábamos anteriormente, en el caso del locus GNAS (y del propio gen GNAS), cada uno de los posibles transcritos (en función de qué exón 1 alternativo se transcriba) presentan fenómeno de imprinting y esta inactivación de uno de sus dos alelos varía según el transcrito (fig. 3). Así:
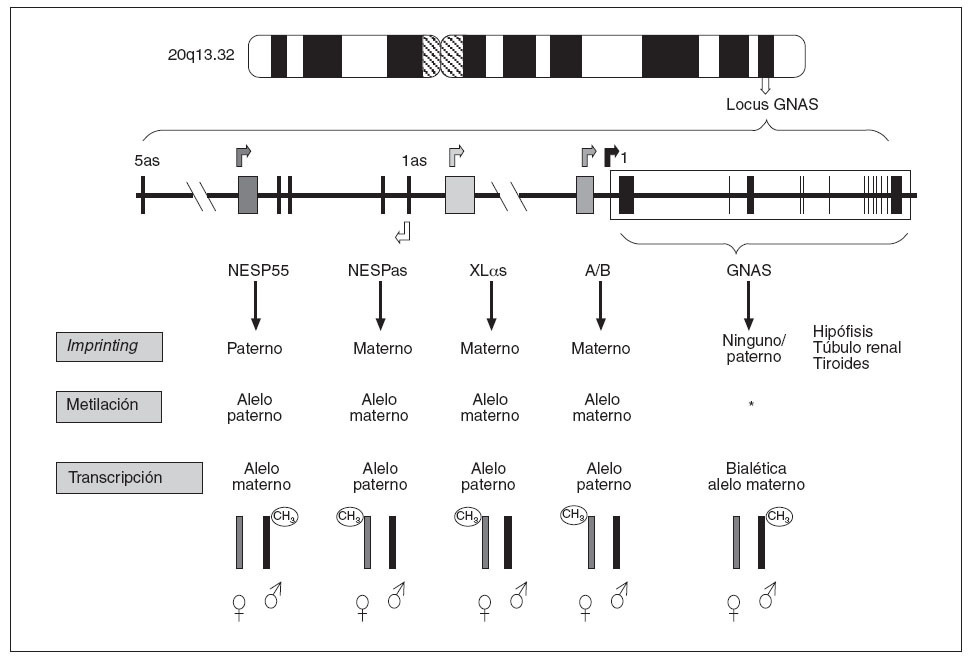
Fig. 3. Patrones de impronta del locus GNAS. El transcrito NESP55 tiene imprinting paterno, mientras que los exones XL αs, A/B y NESPas presentan imprinting materno en todos los tejidos. Los transcritos de Gs α se expresan bialélicamente, excepto en algunos tejidos como el túbulo renal proximal, tiroideas, gónadas e hipófisis. *Se desconoce el mecanismo de impronta del exón 1 de GNAS.
– El transcrito que procede de la unión del exón 1 con el resto de los exones comunes (2-13), que codifica para la propia proteína Gsµ, tiene imprinting paterno (sólo se expresa el alelo materno) en algunos tejidos (hipófisis, tiroides, gónadas y túbulo renal)3,13, pero es de expresión bialélica en el resto de los tejidos. También, el transcrito NESP55 está metilado únicamente en el alelo paterno (en este caso siempre en todos los tejidos), por lo que NESP55 sólo se transcribe a partir del alelo materno9. – Por el contrario,el transcrito A/B tiene imprinting materno (está metilado el alelo heredado de la madre) en todos los tejidos y sólo se transcriben los ARNm del exón A/B del alelo paterno8. De forma similar, la proteína XLµs tiene imprinting materno por lo que sólo se transcribe el alelo paterno9,10 (fig. 3).
Según esto, si consideráramos hipotéticamente como la expresión genética de dos alelos la equivalente al 100% (para cualquier gen), en el caso de la expresión normal de un individuo de los genes A/B, NESP55,XLµs, y en algunos tejidos también GNAS, sería del 50% (un solo alelo).
Los mecanismos que controlan los distintos patrones fisiológicos de imprinting en esta región del genoma no son totalmente conocidos. A partir de datos de estudios de pacientes con seudohipoparatiroidismo (que veremos más adelante), se observa que en un área situada antes del gen GNAS (gen STX16) puede haber un mecanismo capaz de controlar la metilación del exón A/B, ya que deleciones de 3 kb o 4,4 kb en esa región se asocian a veces a trastornos de la metilación A/B y, consecuentemente, a seudohipoparatiroidismo. Por otra parte, y dado que los exones NESP55 y XLµs tienen imprinting opuesto en todos los tejidos, es posible que esta situación esté regulada de forma coordinada. En este sentido, algunos autores señalan que NESPas, un transcrito antisentido que atraviesa el exón NESP55 en dirección opuesta (fig. 3), pudiera regular el silenciamiento o imprinting del exón de NESP55 en el alelo paterno, como recientemente se ha evidenciado14. De hecho, como aval de esta propuesta es importante mencionar que se han descrito transcritos antisentido en relación con varios genes improntados como posibles reguladores de dicha metilación15.
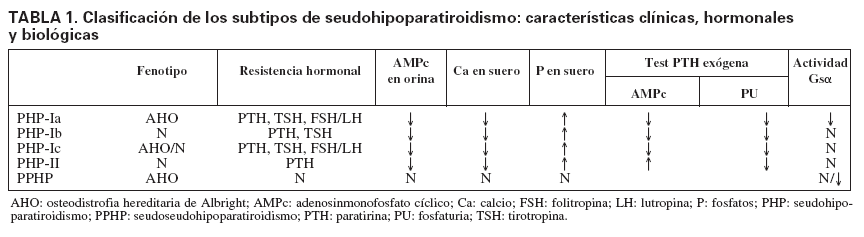
Tipos de seudohipoparatiroidismo y alteración molecularSe distinguen dos formas clínicas de PHP, el tipo I (PHP-I) y el tipo II (PHP-II), según la respuesta a la administración exógena de PTH medida en producción de AMPc urinario (tabla 1). En los pacientes con PHP-I no hay respuesta de aumento de AMPc, mientras que en el PHP-II la respuesta es equivalente a la de un individuo normal16. Sin embargo, la fosfaturia es deficiente en ambos PHP.
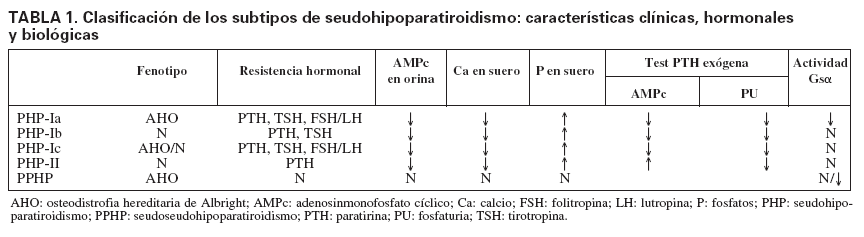
TABLA 1. Clasificación de los subtipos de seudohipoparatiroidismo: características clínicas, hormonales y biológicas
El PHP-I puede ser clasificado a su vez en tres sub-tipos, basados, además de la resistencia a la PTH17, en: a) las características dismórficas de osteodistrofia hereditaria de Albright (AHO); b) la presencia o ausencia de anomalías endocrinas adicionales, y c) la deficiencia en la actividad de la subunidad alfa de la proteína G estimuladora (Gs), medida a través de la respuesta de AMPc (in vitro). La AHO es un síndrome con una amplia variedad de manifestaciones fenotípicas, incluidos estatura baja, obesidad, cara redondeada, osificaciones ectópicas, braquidactilia, que afecta principalmente al cuarto y el quinto metacarpiano, y retraso mental.
Seudohipoparatiroidismo tipo Ia (PHP-Ia) y seudoseudohipoparatiroidismoLos pacientes con AHO y resistencia a la PTH y a otros sistemas hormonales, fundamentalmente a la TSH, son diagnosticados clínicamente como PHP-Ia18. Estos pacientes tienen actividad de proteína G en la mayoría de los tejidos del 50%. Hablamos de seudoseudohipoparatiroidismo (PPHP) cuando, en familias con seudohipoparatiroidismo, los pacientes tienen solamente fenotipo de AHO sin resistencia hormonal (sin hipocalcemia ni hiperfosfatemia y con PTH nor-mal)19.
En cuanto al aspecto molecular, en 1990, Pattern et al20 detectaron y describieron la primera mutación en heterocigosis en el gen que codifica para la subunidad alfa de la proteína Gs (Gsµ) en relación con el cuadro clínico de PHP, y desde entonces se han identificado más de 120 mutaciones diferentes21-27. Estos pacientes, generalmente, presentan mutaciones inactivantes en heterocigosis en uno de los 13 exones o en las regiones de corte y empalme de GNAS y, por lo tanto, presentan herencia autosómica dominante. Aparte de mutaciones puntuales, también se ha descrito a 2 pacientes con AHO que presentan deleciones en el cromosoma28. Asimismo, como indicábamos anteriormente, los pacientes con PHP-Ia y PPHP se encuentran en las mismas familias y estos últimos presentan las mismas mutaciones de GNAS que sus familiares con PHP-Ia19. Un paciente con mutación en el gen GNAS tendrá PHP-Ia si hereda la mutación de la madre, mientras que presentará un fenotipo de PPHP si la misma mutación es heredada del padre3,29,30. Esto es debido al efecto del imprinting paterno que, como hemos visto, afecta al gen que codifica la proteína Gsµ(gen GNAS) en determinados tejidos (fig. 4), según el cual, en condiciones normales, el gen de la proteína Gsµprocedente del padre está fisiológicamente silenciado (no se expresa) en hipófisis, tiroides, gónadas y túbulo renal (por lo tanto, una persona normal funciona en esos tejidos sólo con la carga genética del alelo materno o sea, que podríamos decir, con el 50%), mientras que la expresión del gen GNAS es bialélica en el resto de los tejidos (alelo materno y paterno, o sea con el 100%). En este sentido, cuando hay una mutación en el alelo que se hereda del padre, como su alelo está silenciado en hipófisis, gónadas, tiroides y túbulo renal, la mutación en estos tejidos no tiene efecto clínico en ellos (sigue funcionando la carga del 50% de la madre) y sólo detectaremos alteraciones fenotípicas derivadas de los tejidos en que había expresión bialélica (en este caso, ante la mutación paterna, sólo funciona el alelo materno, o sea tendrá una cargagenética en el 50% en el resto de los tejidos). Éste es el fenotipo característico del seudoseudohipoparatiroidismo. Sin embargo, cuando la mutación se hereda en el alelo materno, en hipófisis, tiroides, gónadas y tú-bulo renal ese alelo está alterado y, al estar el alelo paterno fisiológicamente silenciado por el efecto del imprinting, la expresión de proteína Gsµen esos tejidos está ausente (0%), y en el resto (en los que fisiológicamente hay expresión bialélica), se funcionará con el alelo paterno (50%) y clínicamente se manifestará como un seudohipoparatirodismo-Ia.
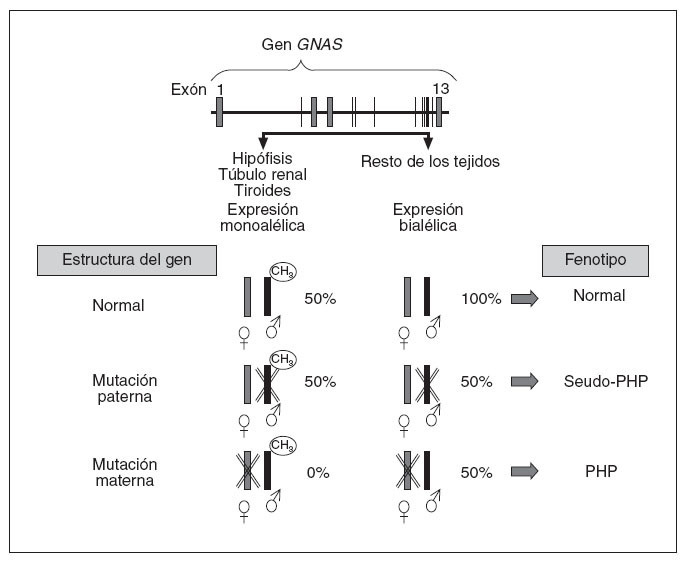
Fig. 4. El gen de la proteína G αs procedente del padre está sometido al fenómeno de imprinting en el túbulo renal proximal, tiroideas, gónadas e hipófisis y no se expresa. En el resto de los tejidos la expresión es bialélica. Cuando el alelo de la madre presenta una mutación, en túbulo renal, tiroides, gónadas e hipófisis, no se expresa ningún alelo (0%), ya que el alelo paterno está inactivado por el efecto del imprinting, y en el resto de los tejidos, como no está inactivado el alelo paterno (es expresión bialélica), se produce el 50% de la actividad. Desde un punto de vista clínico conllevará la aparición de seudohipoparatiroidismo (PHP) en la descendencia. Si es el alelo paterno el mutado, no habrá resistencia hormonal, ya que en hipófisis, tiroides, túbulo renal, etc., es el alelo mutado el que está inactivo (se mantiene el 50% de la actividad correspondiente al alelo materno), y en el resto de los tejidos, en los que debería haber expresión bialélica, un alelo está alterado por la mutación (expresión del 50%), que da un fenotipo de osteodistrofia hereditaria de Albright característica de PHP.
Según todo esto, generalmente cuando tenemos un PHP-Ia y éste es debido a una mutación en el gen GNAS, si no es de novo, habrá sido transmitida por la madre, y ella tendrá un PHP si la recibió de su madre, o un PPHP si la recibió de su padre3,29,30.
Seudohipoparatiroidismo tipo Ic (PHP-Ic)Pacientes que presentan fenotipo AHO y resistencia multihormonal, pero sin evidencias de deficiencia de actividad de Gsµen experimentos in vitro, se clasifican como PHP-Ic3,23. Estos pacientes también pueden presentar mutaciones en GNAS31. Hay una cierta controversia en la comunidad científica respecto a la existencia real del PHP-Ic, dado que la única manera de diferenciar PHP-Ia y PHP–Ic se centra en el estudio in vitro de la actividad de Gsµ. En este sentido, algunos autores postulan que la evaluación de dicho parámetro no plasma la situación real del individuo, y que los pacientes con PHP-1c son en realidad PHP-Ia cuya actividad no puede ser determinada por problemas con la técnica.
En resumen, desde el punto de vista molecular, diremos que los pacientes con PHP-Ia y PHP-Ic presentan, por lo general, mutaciones en heterocigosis en cualquiera de los trece exones codificantes para la proteína Gsµ. Estas alteraciones incluyen mutaciones de cambios de nucleótidos, deleciones e inserciones que afectan a los exones, intrones o lugares de corte y empalme. Incluso se han descrito deleciones del gen completo.
De cara al consejo genético, hemos de tener en cuenta varias premisas: a) cualquier mutación en uno de los dos alelos de un gen puede ser transmitida en el 50% de los casos; b) si la mutación es transmitida por un varón a su descendencia, los hijos presentarán un fenotipo AHO pero sin alteraciones hormonales, es decir, presentarán un cuadro de seudoseudohipoparatiroidismo, y c) sin embargo, si es una mujer la que transmite la mutación, la descendencia presentará seudohipoparatiroidismo (es decir, fenotipo de Albright en presencia de alteraciones hormonales como PTH, TSH y gonadotropinas, etc.). Además, en una familia con PHP, en el caso de un paciente con PPHP, si es varón, en el 50% de los casos transmitirá la mutación que le produjo el PPHP, y los hijos que la hereden tendrán PPHP. Sin embargo, si el paciente con PPHP es una mujer, en el 50% de los casos transmitirá la mutación, y esos hijos que la hereden tendrán un PHP. Tenemos que tener presente que el fenómeno de inactivación fisiológica de un alelo (imprinting) es un proceso dinámico en la transmisión a las generaciones siguientes y en función de quién lo transmita, sea el progenitor varón o mujer, irá inactivándose o activándose en las distintas generaciones.
Seudohipoparatiroidismo tipo Ib (PHP-Ib)La resistencia a PTH aislada, sin AHO, es referida como PHP-Ib3. Algunos casos de PHP-Ib también presentan resistencia moderada a la TSH, pero no suelen presentar resistencia a otras hormonas32,33. Desde el punto de vista clínico podríamos diferenciar dos tipos de PHP-Ib, aquellos con un patrón de herencia autosómica dominante (AD-PHP-Ib), con PHP-Ib en varias generaciones, y otros PHP-Ib esporádicos (casos aislados en una familia). A diferencia de los pacientes con PHP-Ia y PPHP, los pacientes con PHP-Ib presentan actividad normal de Gsµen células de sangre y fibroblastos y, por lo tanto, se esperaba que el defecto molecular estuviese causado por defectos en otro gen distinto de GNAS34. Sin embargo, en una búsqueda a lo largo de todo el genoma en pacientes con PHP-Ib se ha observado que esta afección también está asociada al locus GNAS8,35. En los casos con patrón AD-PHP-Ib, si bien la secuenciación del gen GNAS no mostró ninguna alteración, la mayoría de los pacientes presentaron pérdida en la metilación (con la consecuente pérdida del imprinting o de la inactivación de uno de los dos alelos) de forma exclusiva del exón A/B. Por el contrario, los casos esporádicos de PHP-Ib (sin historia familiar) presentaban un trastorno de la metilación más amplio, que afectaba, además del exón A/B, a los otros exones alternativos (XLµs, NESP55)8,34,36 (véase información anterior sobre la estructura del locus).
Varios estudios han demostrado que los pacientes con AD-PHP-Ib, portadores de la pérdida de metilación aislada para el exón A/B presentan, asimismo, una deleción de 3 kb o 4,4 kb en el gen STX1636-38. Estos hallazgos indican que posiblemente en esta región delecionada hay un elemento regulador del imprinting, necesario para el mantenimiento de la correcta metilación del exón A/B. En las formas esporádicas, en las que la pérdida de metilación es más amplia, no se han detectado deleciones de elementos que regulen el defecto de imprinting. No obstante, es posible que haya otros elementos reguladores en esta región, ya que en dos familias con PHP-Ib, en las que se detectó una pérdida de metilación más amplia, se han encontrado deleciones en un área que incluye el exón NESP55 y el exón antisentido NESPas14.
En cuanto a la patogenia, la pérdida del imprinting conllevaría un aumento de transcripción de dicha región, debido a que el alelo materno que normalmente está metilado deja de estarlo, lo que repercute de alguna forma en la correcta transcripción de GNAS (y de la proteína Gsµ)3.
Estudios genéticos recientes de nuestro grupo han identificado que pacientes con diagnóstico clínico de PHP-Ia presentan alteraciones genéticas similares a los pacientes con AD-PHP-Ib (trastornos en la metilación, en vez de mutaciones puntuales)39. Estos hallazgos han sido corroborados por grupos independientes40, por lo que se puede hablar de un solapamiento molecular de ambos diagnósticos clínicos.
En resumen, podríamos decir que en los pacientes con seudohipoparatiroidismo tipo Ib o en aquellos que no tienen alteraciones en la secuencia las alteraciones en el patrón de metilación, y consecuentemente la pérdida de imprinting, pueden ser las causas del cuadro clínico. Estas alteraciones pueden ser:
– Pérdida aislada en la metilación del exón A/B materno. En algunas familias la pérdida de metilación en este exón se ve acompañada de una deleción en la región del gen STX16 que se hereda de la madre. – Pérdida en la metilación combinada del exón A/B y de XL1µs. Unas pocas familias con esta pérdida de metilación combinada han presentado una deleción (también heredada de la madre) en la región de NESP55 y NESPas.
En un PHP por pérdida de metilación, de cara al consejo genético, podríamos indicar que cuando ésta se relaciona con una deleción de STX16 o NESP55 hay un 50% de riesgo de pérdida de metilación en la generación siguiente (debido a que en el 50% de los casos se transmitirá la deleción), siempre que la deleción se transmita en el alelo materno (el alelo materno normalmente está metilado y, en este caso, al transmitirse la deleción, se transmite sin metilación, o sea, hay doble carga). Por el contrario, si la deleción la transmite el padre, como esa región en el alelo paterno está normalmente sin metilar, no hay cambios en la metilación con respecto a la normalidad (el alelo paterno está sin metilar y el materno está metilado). No obstante, sí que puede tener consecuencias en generaciones siguientes, en las que siempre el porcentaje de riesgo de transmisión de deleciones será del 50% y tendrá relación con pérdida de imprinting cada vez que sea transmitida por mujeres.
En algunos casos, en los que hay pérdida de metilación del exón A/B o de la forma combinada exón A/B más XLµs y no se detectan las deleciones descritas, el consejo genético no puede definirse. En este caso no podemos precisar el riesgo de transmisión de la enfermedad, sólo podemos decir que la enfermedad, en el individuo estudiado, está causada por la pérdida de metilación, pero el riesgo en las generaciones siguientes es incierto.
En definitiva, podemos decir que la genética relacionada con el seudohipoparatiroidismo tipo I puede incluir, como hemos visto, alteraciones en la secuenciación del gen GNAS o en los patrones de metilación de los exones alternativos. Es importante tener siempre bien presente que esta región está sometida al fenómeno de imprinting de cara al correcto diagnóstico del paciente y de sus familiares.
Correspondencia: Dr. L. Castaño. Grupo de Investigación en Endocrinología y Diabetes. Hospital de Cruces. Plaza de Cruces, s/n. 48903 Barakaldo. Bizkaia. España. Correo electrónico: lcastano@osakidetza.net
Manuscrito recibido el 29-7-2008 y aceptado para su publicación el 10-9-2008.
