







El feocromocitoma es un tumor productor de catecolaminas que procede de las células cromafines del sistema nervioso simpático. El 80-85% se localiza en la médula adrenal y el resto, en el tejido cromafín extraadrenal (paraganglioma). Los feocromocitomas suponen el 6,5% de los incidentalomas suprarrenales. Pueden ser esporádicos o encontrarse asociados a varias enfermedades genéticas: neoplasia endocrina múltiple 2, enfermedad de von Hippel-Lindau, neurofibromatosis de tipo 1 y paraganglioma familiar con mutaciones en la succinato deshidrogenasa. El diagnóstico de feocromocitoma debe establecerse bioquímicamente mediante la determinación de metanefrinas plasmáticas y/o urinarias. El diagnóstico de localización inicialmente debe basarse en la realización de tomografía computarizada o resonancia magnética. La gammagrafía con 123I-metayodobencilguanidina (MIBG) es la prueba funcional de imagen de elección; en su defecto puede realizarse con 131I-MIBG. La tomografía por emisión de positrones con 18F-F-fluorodopamina ha resultado de utilidad en enfermedad metastásica. El tratamiento de elección es la cirugía por vía laparoscópica, después de la realización del bloqueo alfaadrenérgico. Aproximadamente el 10% de los tumores son malignos. Para la enfermedad inoperable puede utilizarse la quimioterapia. El pronóstico es bueno, excepto en los casos de enfermedad maligna donde la tasa de supervivencia a los 5 años es menor del 50%. El conocimiento de nuevos genes causantes de enfermedad hereditaria ha supuesto un cambio en las recomendaciones sobre la necesidad de realizar estudio genético.
Pheochromocytomas are catecholamine-secreting tumors that arise from chromaffin cells of the sympathetic nervous system. In 80-85% of cases, these tumors are located in the adrenal medulla while the remainder is located in extra-adrenal chromaffin tissues (paragangliomas). Pheochromocytomas account for 6.5% of incidentally discovered adrenal tumors. These tumors may be sporadic or the result of several genetic diseases: multiple endocrine neoplasia type 2, von Hippel-Lindau syndrome, neurofibromatosis type 1, and familial paraganglioma associated with mutations in succinate dehydrogenase subunits. Diagnosis of pheochromocytoma should first be established biochemically by measuring plasma free metanephrines and urinary fractionated metanephrines. The radiological imaging tests of choice are computed tomography (CT) or magnetic resonance imaging (MRI). The first-line specific functional imaging test is scintigraphy with 123I-metaiodobenzylguanidine (MIBG); if this test is unavailable, scintigraphy with 131I-MIBG is the second choice. Positron emission tomography (PET) with 18F-F-fluorodopamine (F-DA) is useful in metastatic disease. The treatment of choice is laparoscopic surgery after adequate alpha adrenergic blockade. Approximately 10% of tumors are malignant. Chemotherapy is used for inoperable disease. Prognosis is good except in malignant disease, in which 5-year survival is less than 50%. The identification of the genes causing hereditary pheochromocytoma has led to changes in the recommendation for genetic testing.
El feocromocitoma es un tumor productor de catecolaminas que procede de las células cromafines del sistema nervioso simpático. Habitualmente deriva de la médula adrenal. Los feocromocitomas de localización extraadrenal se denominan paragangliomas y pueden originarse en cualquier lugar donde exista tejido cromafín: a lo largo de la cadena ganglionar simpática paraaórtica, en el órgano de Zuckerkandl (en el origen de la arteria mesentérica inferior), en la pared de la vejiga urinaria y en la cadena ganglionar simpática en cuello o mediastino1. La distinción entre un feocromocitoma verdadero y un paraganglioma es importante debido al diferente comportamiento en cuanto al riesgo de malignidad, la posibilidad de otras neoplasias asociadas y la necesidad de estudios genéticos.
La incidencia de feocromocitoma se estima entre 1 y 2 cada 100.000 habitantes y año, y es un 0,3-1,9% de las causas secundarias de hipertensión arterial en la población general. Constituye una causa frecuente de incidentaloma suprarrenal, el 6,5% de dichos tumores2.
Es importante sospechar, confirmar, localizar y resecar el feocromocitoma por varias causas:
- –
La hipertensión arterial asociada es curable con la resección quirúrgica del tumor.
- –
Hay riesgo de muerte súbita.
- –
Por lo menos un 10% de los tumores son malignos.
- –
La detección en los casos de afección familiar puede resultar en el diagnóstico precoz de otros miembros de la familia.
Los avances en el conocimiento del metabolismo de las catecolaminas, el desarrollo de técnicas bioquímicas con altas sensibilidad y especificidad para la detección de estas sustancias en los fluidos biológicos, así como el desarrollo de técnicas de localización no invasivas, han supuesto un avance para el diagnóstico y el tratamiento del feocromocitoma. Simultáneamente, el conocimiento de las distintas presentaciones clínicas del feocromocitoma, el descubrimiento de nuevos fármacos antihipertensivos y el avance de las técnicas anestésicas y quirúrgicas han modificado la aproximación diagnóstica y terapéutica originando una importante disminución en la morbilidad y mortalidad de ésta enfermedad. En los últimos años ha habido un importante progreso en la identificación de la etiología genética del síndrome del paraganglioma familiar. La mayoría de los casos se deben a mutaciones en tres genes (SDHB, SDHC y SDHD), subunidades codificadoras de la succinato deshidrogenasa (SDH) o complejo mitocondrial II. La SDH tiene importantes funciones en el ciclo de Krebs y la cadena respiratoria. El estudio de familias con las distintas mutaciones ha hecho posible estudiar las diferencias entre las presentaciones clínicas según la mutación hallada en cada caso. Las correlaciones fenotipo-genotipo describen la asociación entre una mutación específica y un comportamiento clínico particular.
PRESENTACIÓN CLÍNICALa edad de mayor incidencia se sitúa entre la cuarta y la quinta década de la vida. Afecta a ambos sexos de manera similar. Estos tumores son raros en niños, y cuando ocurren, pueden ser múltiples y asociados a síndromes hereditarios.
La tríada clínica clásica consiste en cefalea (80%), palpitaciones (64%) y diaforesis (57%). Esta tríada en un paciente con hipertensión arterial tiene una sensibilidad para el diagnóstico del 90,9% y una especificidad del 93,8%3.
Cuando los síntomas están presentes, se deben a los efectos farmacológicos del aumento en la concentración de catecolaminas circulantes. La hipertensión resultante puede ser mantenida aproximadamente en la mitad de los pacientes y paroxística en un tercio, mientras el resto de los sujetos presentan normotensión.
La regla del 10 (el 10% son extraadrenales; el 10% en niños; el 10% son múltiples o bilaterales; el 10% recidiva tras la cirugía; el 10% son malignos; el 10% son familiares; el 10% son descubiertos como incidentalomas adrenales) no es del todo cierta. Estudios recientes han demostrado que hasta un 25% son familiares4. También se ha descrito la presencia de feocromocitoma hasta en un 57% de pacientes con incidentaloma adrenal5.
Se debe sospechar un feocromocitoma en las siguientes situaciones: hipertensión resistente al tratamiento, crisis adrenérgicas, historia familiar de feocromocitoma, síndrome genético que predisponga, incidentaloma adrenal radiológicamente compatible, hipertensión en paciente joven y respuesta presora durante la inducción de la anestesia.
DIAGNÓSTICO BIOQUÍMICOEl diagnóstico se confirma típicamente con la determinación de catecolaminas y metanefrinas fraccionadas plasmáticas y urinarias. Sin embargo, hay diferencias en las aproximaciones diagnósticas y todavía no hay un consenso sobre cuál es la prueba más precisa.
En general, los tests bioquímicos más utilizados han sido la determinación de la excreción en orina de 24h de catecolaminas y metanefrinas totales, pero como que la mayoría del metabolismo de las catecolaminas es intratumoral, con formación de metanefrina y normetanefrina, hoy se determinan catecolaminas y metanefrinas fraccionadas6. Los laboratorios utilizan técnicas como cromatografía líquida de alta resolución (HPLC) o espectrometría de masas. Estas técnicas han solventado los problemas que presentaban los análisis fluorimétricos, como los falsos positivos causados por fármacos y contrastes radiológicos.
La determinación de la concentración de metanefrinas plasmáticas se ha propuesto como método de elección7. Se basa en el hecho de que aunque los feocromocitomas pueden segregar catecolaminas sólo episódicamente, las metabolizan de forma continua. De esta forma la concentración de estas sustancias se mantiene elevada permanentemente en presencia de un tumor, incluso aunque la liberación de catecolaminas sea paroxística.
La eficacia diagnóstica de las distintas determinaciones bioquímicas para la detección del feocromocitoma se valoró en un estudio multicéntrico7, que incluyó a 1.003 pacientes. Fueron estudiados por sospecha clínica de feocromocitoma o como parte del cribado de síndrome familiar.
Se compararon las determinaciones plasmáticas de metanefrinas con las de las demás pruebas diagnósticas: catecolaminas plasmáticas y determinaciones urinarias de metanefrinas totales y fraccionadas, catecolaminas y ácido vanilmandélico (VMA).
Las pruebas que mostraron mayor sensibilidad fueron las metanefrinas plasmáticas (99%), seguidas de las metanefrinas urinarias fraccionadas (97%), con diferencias estadísticamente significativas respecto a las demás determinaciones (catecolaminas urinarias, 86%; catecolaminas plasmáticas, 84%, y metanefrinas urinarias totales, 77%). La determinación menos sensible fue la de VMA (64%). Se observó mayor sensibilidad de todos los tests en la detección del feocromocitoma esporádico respecto al diagnóstico del tumor hereditario. Sin embargo, la especificidad fue superior en todas las determinaciones para el feocromocitoma hereditario. Ofreció la mayor especificidad la determinación de VMA (95%), seguida de las metanefrinas totales urinarias (93%), las metanefrinas plasmáticas (89%), las catecolaminas urinarias (88%), laas catecolaminas plasmáticas (81%) y las metanefrinas fraccionadas urinarias (69%). A la vista de estos datos, concluyeron que la determinación de metanefrinas plasmáticas es la prueba de primera elección en el diagnóstico de feocromocitoma.
Sawka et al8, al comparar la eficacia diagnóstica entre la determinación de metanefrinas plasmáticas fraccionadas y la combinación de metanefrinas totales más catecolaminas urinarias en 349 pacientes estudiados por presentar un cuadro clínico indicativo, incidentaloma adrenal o historia familiar, encontraron una sensibilidad del 97 y el 90% respectivamente, con especificidad del 85 y el 96%. Con base en la obtención de un 15% de falsos positivos en la determinación de metanefrinas plasmáticas, concluyeron que la determinación de metanefrinas plasmáticas puede ser el test de elección en los pacientes de alto riesgo. Similares resultados mostró un estudio posterior en el que se incluyó a 1.028 pacientes para comparar las determinaciones de metanefrinas plasmáticas y catecolaminas urinarias fraccionadas9. Según las recomendaciones derivadas del Primer Simposium Internacional sobre el feocromocitoma, la determinación de las concentraciones de metanefrinas urinarias o plasmáticas es más precisa que la de catecolaminas, y los intervalos de referencia de normalidad deben favorecer a la sensibilidad frente a la especificidad para evitar los falsos negativos en el diagnóstico10.
Se ha descrito también que el ajuste en la interpretación de los resultados obtenidos en la determinación de metanefrinas plasmáticas fraccionadas según la edad de los pacientes puede disminuir significativamente la aparición de resultados falsos positivos en la detección del feocromocitoma esporádico. La fracción de normetanefrina plasmática suele ser en la mayoría de los casos la causa de los falsos positivos, y aumenta con la edad11.
El metaanálisis de las determinaciones de metanefrinas plasmáticas realizado con el fin de comparar los resultados de los diversos estudios mostró una sensibilidad entre el 96 y el 100% y una especificidad que oscilaba entre el 85 y el 100%.
Concluyen los autores que el hallazgo de concentraciones de metanefrinas plasmáticas dentro del rango de referencia excluye la posibilidad de feocromocitoma, pero un test positivo aumenta la sospecha sin confirmar el diagnóstico12.
En general todas las pruebas mostraron una menor sensibilidad y mayor especificidad para el diagnóstico de feocromocitoma hereditario que para el del esporádico7. Es probable que esto tenga relación con el menor tamaño que suelen presentar los tumores en los síndromes familiares. Por el contrario, los pacientes con feocromocitoma esporádico suelen presentar tumores más grandes y cuadros clínicos más indicativos de hipersecreción adrenérgica.
Para confirmar el diagnóstico, el resultado de las determinaciones hormonales debe ser por lo menos el doble del límite superior del rango de referencia13. De ser posible, el paciente debe abandonar 2 semanas antes cualquier medicación. Sin embargo, si el paciente precisa continuar con tratamiento antihipertensivo, los diuréticos, los inhibidores de la enzima de conversión de angiotensina (IECA), los antagonistas del calcio, el minoxidil y los bloqueadores alfa o beta han mostrado mínima interferencia en las determinaciones.
Existen diversas situaciones clínicas y fármacos que pueden alterar el resultado de las determinaciones de los parámetros estudiados (tablas 1 y 2).
Medicaciones que pueden aumentar las concentraciones de catecolaminas y metanefrinas
| Antidepresivos tricíclicosAntipsicóticosLevodopaDescongestivos nasales, antitusígenos, broncodilatadores (contienen agonistas de receptores adrenérgicos)AnfetaminasBuspironaProclorperazinaReserpinaEtanolParacetamolSupresión brusca del tratamiento con clonidina |
Cuando se miden catecolaminas o metanefrinas urinarias, se debe incluir la determinación de creatinina urinaria para confirmar una recogida adecuada.
La extracción para metanefrinas plasmáticas debe realizarse con el paciente en posición supina durante 30min, ya que se ha identificado que la sedestación es un factor que puede dar lugar a resultados falsos positivos con mayor probabilidad10.
Otras pruebas diagnósticasCatecolaminas plasmáticas: no se aconseja su utilización por su escasa precisión para el diagnóstico.
VMA: su determinación en orina de 24h tiene baja sensibilidad respecto a las metanefrinas urinarias.
Neuropéptido Y: sus concentraciones están elevadas en el 87% de los pacientes con feocromocitoma, pero también carece de la fiabilidad que ofrecen las determinaciones de catecolaminas y metanefrinas. Los paragangliomas no suelen producir NPY generalmente.
Cromogranina A: la familia de las cromograninas, cuyos miembros más conocidos son las cromograninas A (CgA) y B y la secretogranina II, han mostrado ser determinaciones diagnósticas eficaces. La CgA es almacenada y liberada desde la médula adrenal y de las vesículas de las neuronas simpáticas. La demostración inmunohistoquímica de CgA en un tumor adrenal es indicio firme de feocromocitoma. La CgA ha sido descrita como el marcador general más preciso de tumores neuroendocrinos. Se han realizado estudios para valorar su papel en el seguimiento de pacientes con feocromocitoma en comparación con las determinaciones urinarias de catecolaminas y metanefrinas, y se ha observado una sensibilidad del 100% con la determinación de catecolaminas urinarias más CgA, frente al 86% que presenta su determinación aislada14. El papel de la CgA en el diagnóstico diferencial entre feocromocitomas malignos y benignos es controvertido; mientras que algunos autores han descrito concentraciones más elevadas en tumores malignos, otros no han encontrado diferencias15. Las concentraciones de CgA se correlacionan con el tamaño del feocromocitoma y la extensión de la enfermedad13. Este hecho puede explicar la normalidad del marcador tumoral en los casos de feocromocitoma hereditario y lesiones de pequeño tamaño. Sin embargo, la elevación de CgA no es específica de feocromocitoma y puede estar elevada en otros tumores neuroendocrinos. La insuficiencia renal y la ingesta de inhibidores de la bomba de protones producen aumento en las concentraciones de CgA circulante. Su utilidad en el seguimiento de los feocromocitomas intervenidos ha sido estudiada. Se ha objetivado una rápida normalización de los valores de CgA, a pesar de la persistencia de excreción aumentada de catecolaminas posquirúrgica durante 2 o 3 semanas tras la cirugía, lo que indica un posible papel de este marcador en la demostración precoz de curación en los pacientes intervenidos de feocromocitoma13.
EM66: El péptido derivado de la secretogranina II EM66 se genera en tumores cromafines. Se ha observado su aumento en los feocromocitomas, más en tumores benignos16, y se ha descrito como un marcador muy sensible para el diagnóstico y el seguimiento de estos tumores17.
Enolasa neuroespecífica (NSE): marcador de tumor neuroendocrino con menor sensibilidad y especificidad que la CgA.
Metoxitiramina: metabolito de la dopamina. Se ha descrito la utilidad de su determinación plasmática en tumores productores de dopamina en comparación con la dopamina urinaria. No obstante su papel puede estar restringido debido a la baja frecuencia de presentación de estos tumores18.
Prueba de supresión con clonidina: se ha utilizado con el fin de detectar los falsos positivos en las determinaciones plasmáticas de metanefrinas y catecolaminas. La clonidina es un agonista alfa-2 de acción central que en condiciones normales suprime la secreción de catecolaminas. La persistencia de concentraciones de catecolaminas o metanefrinas elevadas tras su administración confirma la autonomía tumoral. La respuesta normal 3h tras una dosis de 0,3mg es una disminución respecto a la basal en la concentración de noradrenalina plasmática > 50% o la de normetanefrina > 40%. Deben suprimirse con anterioridad los tratamientos con diuréticos, bloqueadores beta y antidepresivos tricíclicos y no realizarse en pacientes hipovolémicos, con el fin de evitar un cuadro de hipotensión severa. El tratamiento con bloqueadores alfa no interfiere con el resultado de la prueba19.
Pruebas de provocación: debido a los avances en las técnicas de laboratorio empleadas para la determinación de catecolaminas y metanefrinas, rara vez se precisa su utilización12.
En resumen, el clínico tiene varias opciones cuando se trata de diagnosticar un feocromocitoma. La elección de uno u otro test debe considerarse en función del grado de sospecha clínica. En los pacientes con alto riesgo (tabla 3), la determinación de metanefrinas plasmáticas puede estar justificada por su elevada sensibilidad. Sin embargo, en la mayoría de las ocasiones en que el diagnóstico es poco probable, la utilización de un parámetro que ofrezca mayor especificidad con aceptable sensibilidad, como la determinación de metanefrinas urinarias, puede ser de elección para evitar la alta tasa de falsos positivos en una población de bajo riesgo11,20.
DIAGNÓSTICO DE LOCALIZACIÓNDebe realizarse una vez confirmado el diagnóstico bioquímico. La tomografía computarizada (TC) y la resonancia magnética (RM) deben ser utilizadas como técnicas de localización en primer lugar. Además deben realizarse sólo del abdomen inicialmente. Aproximadamente, entre el 9 y el 23% de los tumores son extraadrenales3,21, pero el 95% se localiza en el abdomen y la pelvis22. Las localizaciones extraadrenales más frecuentes son las áreas paraaórticas abdominales (75%), la vejiga (10%), el tórax (10%), la cabeza y el cuello (3%) y la pelvis (2%)23. Ambas técnicas poseen alta sensibilidad (el 98 y el 100% respectivamente), pero escasa especificidad (el 70 y el 67%)3 por la alta prevalencia de los incidentalomas adrenales.
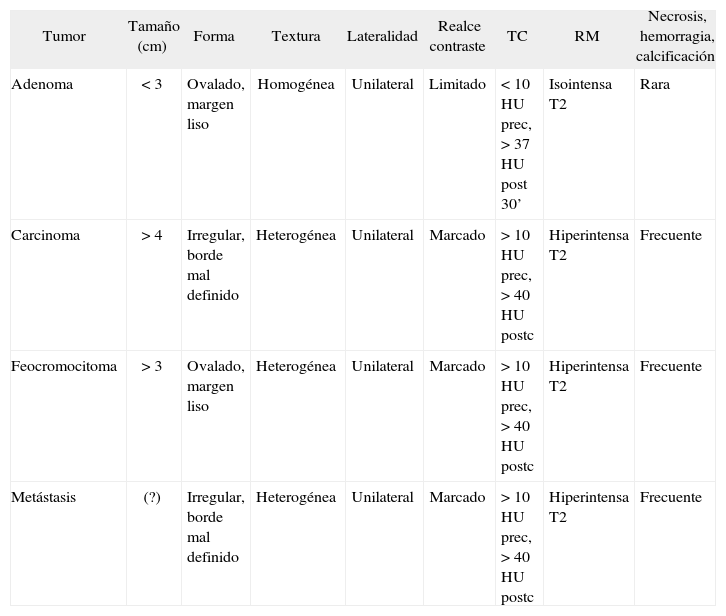
Tomografía computarizadaSe pueden detectar tumores a partir de 0,5cm de diámetro, para lo que deben realizarse secciones de 2-5mm de grosor. El aumento en la prevalencia de los incidentalomas adrenales hace necesario hallar signos radiológicos que permitan un diagnóstico específico, con el fin de decidir el tratamiento adecuado en cada caso. El coeficiente de atenuación sin contraste expresado en unidades Hounsfield (HU) está siendo utilizado para diferenciar los adenomas adrenales de otros tumores. Esto se basa en el hecho de que el contenido citoplásmico de lípidos es alto en los adenomas, pero escaso en los demás tumores24. Una masa homogénea con una densidad de menos de 10 HU es probable que sea un adenoma, mientras que el diagnóstico es incierto si es más densa25. Cuando los resultados son inconcluyentes, puede administrarse contraste radiológico y valorar el realce de la imagen, así como la desaparición del contraste. El feocromocitoma suele mostrar un valor de atenuación precontraste mayor que los adenomas26, junto con un marcado realce tras la administración de contraste y a menudo, sobre todo en los de mayor tamaño, la imagen es heterogénea, con áreas de necrosis y hemorragia27 (tabla 4).
Características radiológicas de los tumores suprarrenales
| Tumor | Tamaño (cm) | Forma | Textura | Lateralidad | Realce contraste | TC | RM | Necrosis, hemorragia, calcificación |
| Adenoma | < 3 | Ovalado, margen liso | Homogénea | Unilateral | Limitado | < 10 HU prec, > 37 HU post 30’ | Isointensa T2 | Rara |
| Carcinoma | > 4 | Irregular, borde mal definido | Heterogénea | Unilateral | Marcado | > 10 HU prec, > 40 HU postc | Hiperintensa T2 | Frecuente |
| Feocromocitoma | > 3 | Ovalado, margen liso | Heterogénea | Unilateral | Marcado | > 10 HU prec, > 40 HU postc | Hiperintensa T2 | Frecuente |
| Metástasis | (?) | Irregular, borde mal definido | Heterogénea | Unilateral | Marcado | > 10 HU prec, > 40 HU postc | Hiperintensa T2 | Frecuente |
postc: después del contraste; prec: antes del contraste; HU: unidades Hounsfield; RM: resonancia magnética; TC: tomografía computarizada. Tomada de Young, 2006.
Si la TC de abdomen y pelvis es negativa, se debe practicar otras de tórax y cuello28. Para la detección de tumores pequeños es preferible la realización de TC helicoidal. Para las lesiones limitadas a las suprarrenales, la TC con contraste tiene altas sensibilidad y especificidad, que disminuyen si la lesión se localiza fuera de ellas24. Se ha descrito la aparición de crisis hipertensivas con la administración de contraste, por lo que se ha pretratado con bloqueadores alfa a los pacientes con diagnóstico bioquímico probable de feocromocitoma, aunque en este aspecto los estudios son contradictorios29. Si el tumor es localizado, debe realizarse una prueba funcional para confirmar que se trata de un feocromocitoma y descartar enfermedad metastásica. Si es negativo, debe realizarse una RM.
Resonancia magnéticaEs la técnica de elección en niños y mujeres gestantes y otras situaciones que requieran la mínima exposición a la radiación30. En las secuencias en T1 de la RM, el feocromocitoma muestra una señal similar a la del hígado, riñón y músculo y se diferencia con facilidad del tejido graso.
La naturaleza rica en lípidos de los adenomas corticales es de gran ayuda en diferenciar estas neoplasias benignas de los feocromocitomas, carcinomas, metástasis y seudoquistes hemorrágicos31. La hipervascularización del feocromocitoma hace que el tumor aparezca característicamente hiperintenso en la secuencia potenciada en T2, respecto al hígado y al músculo21. Entre las ventajas de la RM se encuentran la alta sensibilidad en detectar enfermedad adrenal y la ausencia de exposición a radiaciones ionizantes. Además, no precisa preparación con bloqueadores adrenérgicos, ya que puede realizarse sin contraste intravenoso, y en caso de ser necesarios, éstos son muy seguros32. Utilizada en combinación con la 123I-metayodobencilguanidina (MIBG), se han descrito sensibilidad y especificidad del 100% para la localización de feocromocitomas33.
EcografíaSu utilidad es muy limitada, por lo que no se recomienda excepto en casos en que no se pueda realizar TC o RM34.
Imágenes funcionalesDado que no hay un consenso sobre la existencia de criterios clínicos, bioquímicos o de imagen que sean capaces de predecir la malignidad o multicentricidad del feocromocitoma, la necesidad de descartar estas situaciones es importante35. Las células del feocromocitoma expresan sistemas de transporte de membrana y vesiculares de catecolaminas, lo que permite imagen con 131I y 123I-MIBG, así como con varios isótopos para tomografía por emisión de positrones (PET)33.
Gammagrafía con MIBGCuando no es posible la localización tumoral con TC o RM36, la gammagrafía con 123I-MIBG puede resultar de utilidad. Esta prueba es la técnica funcional de elección debido a su alta especificidad10. También es válida en la localización de tumores múltiples. La realización de la gammagrafía con MIBG ha sido considerada en ocasiones como superflua cuando la TC o la RM son diagnósticas35, excepto en el caso de tumores adrenales grandes (> 10cm) por la posibilidad de malignidad, enfermedad bilateral o en el supuesto de que se trate de un paraganglioma, ya que en este caso el riesgo de tumores múltiples está aumentado22. La MIBG es un compuesto con cierto parecido a la noradrenalina que es captada por el tejido adrenal; sin embargo, no tiene apenas afinidad por los receptores adrenérgicos. Su captación puede estar alterada por ciertos fármacos: descongestivos nasales, antidepresivos tricíclicos, antagonistas del calcio, labetalol, antipsicóticos y cocaína. A pesar de que el labetalol pueda dar lugar a falsos negativos, otros bloqueadores alfa o beta no interfieren37. La utilización de 123IMIBG es preferible a la de 131I-MIBG, ya que la vida media es más corta y la sensibilidad es mayor (el 83 frente al 77%). Con ambos isótopos la especificidad es cercana al 100%38. Se precisa realizar bloqueo tiroideo previo.
A pesar de las ventajas de la gammagrafía 123IMIBG, su sensibilidad no es la óptima para la detección de metástasis, por lo que en ocasiones es necesaria la realización de PET con con [18F]fluorodopamina ([18F]FDA), [18F]fluorodesoxiglucosa u octreoscan10.
Tomografía por emisión de positronesLa PET se ha realizado con [18F]fluorodesoxiglucosa, [18F]FDA, [18F]fluorodopa, [11C]hidroxiefedrina y [11C]adrenalina. La dopamina es un sustrato específico para el transportador de noradrenalina, comparado con otras aminas, incluidas la noradrenalina y la DOPA. La [18F]FDA es un agente excelente para localizar feocromocitomas adrenales y extraadrenales39, incluidas las lesiones metastásicas40. Hay estudios que indican que la utilización de PET con [18F]FDA supera al uso de 123I-MIBG en la localización de enfermedad metastásica, ofreciendo además varias ventajas: menor radiación, no es necesario el bloqueo tiroideo y mayor rapidez en la realización de la prueba, ya que se realiza inmediatamente después de la administración de [18F]FDA y no 24 a 48h después33. La utilización de [18F]fluorodesoxiglucosa puede ser adecuada para localización de tumores desdiferenciados41, pero no es específica para feocromocitoma, por lo que no debe utilizarse como estudio inicial33,42. La pérdida de los transportadores de los neurotransmisores específicos da lugar a la incapacidad para la acumulación de estos isótopos y la consiguiente falta de localización, por lo que puede ser necesario realizar PET con [18F]fluorodesoxiglucosa u octreoscan33.
OctreoscanSe ha descrito que hasta el 73% de las células del feocromocitoma expresan receptores para la somatostatina (predominantemente tipos 2 y 4)43, al igual que otros tumores neuroendocrinos. Sin embargo, la interpretación del octreoscan se complica por la presencia normal de receptores de somatostatina en una amplia variedad de tejidos. Las infecciones y los procesos inflamatorios pueden dar lugar a falsos positivos44. En general se ha mostrado más eficaz en la localización de enfermedad metastásica37 que en los procesos benignos. La realización de PET o de octreoscan no está recomendada como estudio de localización inicial10.
En la figura 1 se muestra el algoritmo diagnóstico.
TRATAMIENTO MÉDICO DEL FEOCROMOCITOMA.")
El tratamiento de elección del feocromocitoma es la cirugía. La mejoría en el pronóstico de la cirugía del feocromocitoma es multifactorial, e incluye un mejor manejo anestésico, la monitorización perioperatoria y los fármacos antihipertensivos. La tasa de mortalidad ha disminuido de un 20% en 195145 a menos del 5% en la actualidad. La preparación farmacológica preoperatoria es un factor clave en la reducción de la morbilidad y ha sido ampliamente demostrada46, aunque algunos estudios refieren resultados contradictorios en este sentido47. El objetivo del tratamiento preoperatorio consiste en controlar la hipertensión y aumentar el volumen circulante.
Bloqueadores alfaEn ausencia de estudios controlados de gran número de pacientes, la utilización de bloqueadores alfa no específicos como la fenoxibenzamina ha sido el procedimiento habitual. El tratamiento preoperatorio con bloqueadores alfa se utiliza para contrarrestar la liberación masiva de catecolaminas durante la cirugía. Sin embargo, varios estudios han descrito la aparición de crisis hipertensivas durante la manipulación del tumor independientemente de que se hubiera usado el bloqueo alfa previamente o no46.La preparación prequirúrgica durante períodos más largos tampoco se ha mostrado más eficaz. Debe comenzarse con una dosis de 10mg/12h e ir aumentando cada 2-3 días hasta conseguir un control adecuado de la presión arterial. La mayoría de los pacientes requieren entre 80 y 100mg diarios. La utilización bloqueadores alfa-1 como la doxazosina (2-8mg/día), prazosina (2-5mg/8h) o terazosina (2-5mg/día) puede resultar de utilidad para solventar ciertas desventajas derivadas del uso de la fenoxibenzamina. Dado que no producen bloqueo de los receptores alfa-2, estos fármacos no aumentan la liberación de noradrenalina, por lo que no producen taquicardia refleja. Su acción es menos duradera, permite un rápido ajuste de dosis y disminuye la duración de la hipotensión postoperatoria.
Bloqueadores betaUna vez que se ha establecido el bloqueo alfa, puede iniciarse el tratamiento con bloqueadores beta si el paciente presenta arritmia o taquicardia. Un bloqueador beta nunca debe utilizarse en ausencia de bloqueo alfa, ya que puede exacerbar la vasoconstricción inducida por adrenalina inhibiendo su componente vasodilatador, lo que puede lugar a un mayor aumento de la presión arterial con aumento de la poscarga, y es posible la aparición de disfunción miocárdica y edema pulmonar48.
El labetalol, un fármaco con acción antagonista alfa y beta, puede utilizarse en una dosis entre 200 y 600mg cada 12h. La proporción alfa/beta es 1/3, por lo que el bloqueo alfa puede resultar insuficiente.
Antagonistas del calcioHasta la fecha, la utilización de antagonistas del calcio para la preparación de la cirugía del feocromocitoma ha sido descrita en pocos casos; no obstante, han demostrado que son de utilidad en el control de la hipertensión arterial y otros síntomas en pacientes con feocromocitoma49. Los antagonistas del calcio tienen propiedades vasodilatadoras arteriolares, y en concesuencia se reduce la resistencia vascular periférica (inhibición de la liberación de calcio intracelular mediada por noradrenalina) y mejora la función ventricular izquierda. Pueden ser utilizados en combinación con bloqueadores alfa-1 en casos de hipertensión resistente. Además no producen hipotensión ortostática y resultan de utilidad en pacientes con hipertensión paroxística.
MetirosinaEste fármaco inhibe la enzima tirosina hidroxilasa, que es el paso limitante en la síntesis endógena de catecolaminas. De esta forma facilita el control de la presión arterial antes y durante la cirugía, especialmente durante la inducción de la anestesia y la manipulación del tumor, circunstancias en las que se produce la activación simpática y la liberación de catecolaminas50. El tratamiento se comienza con una dosis de 250mg cada 8h, y se aumenta la dosis cada 2-3 días. Este fármaco cruza la barrera hematoencefálica y puede producir efectos secundarios importantes, por lo que debe utilizarse con cuidado y sólo cuando otros agentes hayan sido ineficaces o en pacientes en que se prevé una marcada manipulación o destrucción tumoral.
En resumen, el uso apropiado de antagonistas del calcio y bloqueadores alfa-1 es efectivo y seguro y no presenta los efectos adversos asociados con el bloqueo alfa completo y prolongado de la fenoxibenzamina.
TRATAMIENTO QUIRÚRGICOLa resección por vía laparoscópica es la técnica de elección, en pacientes con tumores menores de 8cm, ya que ofrece varias ventajas respecto a la cirugía convencional51-54. No está indicada cuando hay evidencia preoperatoria de infiltración de los tejidos circundantes55,56.
La adrenalectomia debe ser completa. La cirugía laparoscópica debe convertirse en cirugía abierta en caso de dificultades en la disección, invasión, adherencias o inexperiencia del cirujano34,57. Se ha observado una mortalidad del 2,4% y una morbilidad de 24%58. Las alteraciones hemodinámicas intraoperatorias en pacientes intervenidos de feocromocitoma por vía laparoscópica han resultado ser similares a las producidas en la cirugía abierta59. Sin embargo, en los pacientes sometidos a laparoscopia se registraron menos episodios de hipotensión intraoperatoria y fueron menos severos. La duración de la cirugía fue similar en ambos grupos. La recuperación posquirúrgica fue más temprana, la duración de la hospitalización menor y la recuperación de la actividad física normal más rápida en el grupo de pacientes sometidos a cirugía laparoscópica60. El tamaño tumoral, la duración de la anestesia y las concentraciones previas de catecolaminas urinarias han sido considerados factores independientes de las complicaciones perioperatorias45, aunque existe alguna controversia en este aspecto.
La aparición intraoperatoria de crisis hipertensivas por la liberación de catecolaminas puede ocurrir en cuanto se crea el neumoperitoneo en la cirugía por vía laparoscópica61-63. Se ha descrito la asociación entre la creación del neumoperitoneo y la manipulación tumoral con el aumento marcado en las cifras de catecolaminas circulantes64. Una presión intraabdominal menor durante la adrenalectomía laparoscópica ha demostrado causar menor liberación de catecolaminas y menos alteraciones hemodinámicas65. La introducción de CO2 para crear el neumoperitoneo induce cambios hemodinámicos como resultado de la absorción de CO2. La hipercapnia resultante puede causar liberación de catecolaminas que dé lugar a alteraciones hemodinámicas y arritmias. Para evitar esto se ha propuesto la utilización de helio para la resección por vía laparoscópica del feocromocitoma66. Sin embargo, estudios posteriores han observado que la insuflación de la cavidad peritoneal se asocia con alteraciones hemodinámicas importantes y liberación de catecolaminas, independientemente de que el gas utilizado fuera helio o CO2, y que las cifras hormonales vuelven a la normalidad sólo después de la eliminación del gas67.
Cuando la afección es bilateral, se ha planteado la realización de adrenalectomía bilateral parcial para preservar la función glucocorticoidea, pero se han descrito recidivas en un 10-20%68. En los pacientes con MEN 2, debido a que la enfermedad de la médula adrenal es difusa, se aconseja adrenalectomía bilateral completa cuando la afección sea claramente bilateral. En la enfermedad de von Hippel Lindau, la adrenalectomia parcial puede ser una opción mejor, debido a la diferente afección adrenal.
TRATAMIENTO INTRAOPERATORIOEl tratamiento quirúrgico de los feocromocitomas conlleva un alto riesgo de complicaciones perioperatorias. La manipulación del tumor, así como otros estímulos, puede dar lugar a la aparición de crisis hipertensivas o arritmias, y tras la resección tumoral puede aparecer un cuadro de hipotensión arterial severa. El tratamiento hemodinámico de estos pacientes a veces es complicado debido al fenómeno de down regulation de los receptores adrenérgicos secundario a la exposición prolongada a altas concentraciones de catecolaminas.
La crisis hipertensivas que tiene lugar durante la cirugía se han tratado con nitroprusiato sódico por su rápido comienzo de acción y la corta duración de su efecto12. Es un potente vasodilatador arterial y venoso, que produce disminución de la presión arterial sistólica y diastólica. Se administra en forma de infusión intravenosa a una dosis entre 0,5 y 1μg/kg/min y se ajusta la dosis según la respuesta de la presión arterial. Produce un aumento reflejo de la frecuencia cardiaca, por lo que hasta un 60% de los pacientes requieren agregar tratamiento con bloqueadores beta durante la cirugía, frente a un 20% cuando se utilizan antagonistas del calcio66.
La fentolamina es un bloqueador alfa no selectivo y de acción corta que se administra a una dosis de 1mg en forma de bolo intravenoso, seguido si es necesario de bolos de 5mg. La respuesta máxima se alcanza en 2-3min y dura 10-15min.
El nicardipino puede utilizarse también en forma de infusión intravenosa para control de las crisis hipertensivas. Su efecto en la estabilidad hemodinámica durante la cirugía se ha estudiado en un grupo de 105 pacientes69. Los pacientes recibieron tratamiento con nicardipino durante los días previos a la cirugía (20-60mg/día). Se utilizó una infusión intravenosa de nicardipino para mantener la presión arterial entre 90 y 160mmHg durante el acto quirúrgico. Las crisis hipertensivas intraoperatorias fueron tratadas aumentando el ritmo de infusión.
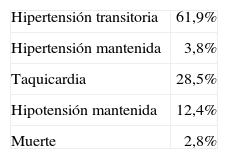
El tratamiento preoperatorio con antagonistas del calcio no previno totalmente la aparición de la inestabilidad hemodinámica, que se produjo en el momento de la intubación en el 17% de los pacientes y en el de la manipulación tumoral en el 40%. Sin embargo, fue eficaz en el control intraoperatorio de la presión arterial. Las complicaciones perioperatorias se exponen en la tabla 5.
Complicaciones perioperatorias en pacientes con feocromocitoma tratados con antagonistas del calcio
| Hipertensión transitoria | 61,9% |
| Hipertensión mantenida | 3,8% |
| Taquicardia | 28,5% |
| Hipotensión mantenida | 12,4% |
| Muerte | 2,8% |
Tomado de Lebuffe G et al69.
Se ha descrito la utilización de los bloqueadores alfa-1 para prevenir las crisis hipertensivas intraoperatorias, por su vida media más corta, con el fin de evitar el bloqueo alfa no selectivo (más prolongado) en casos de hipotensión severa. La utilización de bloqueadores alfa durante la cirugía no previene del todo la aparición de labilidad hemodinámica, y se han descrito63 resultados similares a los obtenidos con antagonistas del calcio.
La utilización de lidocaína o esmolol puede ser de utilidad en caso de aparición de arritmias.
TRATAMIENTO POSTOPERATORIOHipotensión arterialLa reposición de volumen es el tratamiento de elección si aparece un cuadro de hipotensión arterial. Es todavía más importante cuando la preparación prequirúrgica se realiza con fenoxibenzamina o con metirosina. Esto se debe a que la metirosina inhibe la síntesis de catecolaminas en el tumor y en el sistema nervioso central y la fenoxibenzamina bloquea la acción de las catecolaminas. En esta situación el lecho vascular se encuentra en situación de vasodilatación sostenida, por lo que el uso de fármacos presores es más complicado. Durante las 24-48h tras la cirugía, el volumen que se repone suele ser considerable (0,5-1,5 veces el volumen total) y se determina mediante un riguroso control de las constantes hemodinámicas del paciente.
HipoglucemiaAparece en un 10-15% de los pacientes y ocurre porque se suprime la inhibición de la secreción de insulina mediada por catecolaminas70. Se trata con una infusión intravenosa de glucosa.
FEOCROMOCITOMA MALIGNOAlrededor del 10% de los feocromocitomas y hasta un 50% de los paragangliomas son malignos. Histológica y bioquímicamente son similares. La única prueba evidente de la presencia de un feocromocitoma maligno es la invasión local o la presencia de metástasis, que puede ocurrir hasta 20 años después de la resección del tumor71. Se ha señalado que la valoración de una serie de características morfológicas e histológicas de los tumores pueden apoyar el diagnóstico de malignidad72. Los pacientes con la mutación de la succinato deshidrogenasa B tienen más probabilidad de desarrollar enfermedad maligna.
El tratamiento de elección es la exéresis del tumor. La tasa de supervivencia a los 5 años es menor del 50%. Las lesiones óseas pueden tratarse con radioterapia externa o crioablación. El tratamiento con 131IMIBG está considerado hoy en día como el tratamiento más eficaz junto con la cirugía73,74, aunque su valor terapéutico es limitado75-77. Recientemente se han comunicado78 resultados más satisfactorios en pacientes tratados con múltiples dosis de 131I-MIBG, e incluso una mayor tasa de remisiones en pacientes con tumores malignos irresecables tratados con dosis altas entre 557mCi y 1.185mCi de 131I-MIBG79. Se ha descrito una mayor respuesta de los tejidos blandos que con las metástasis óseas. En los pacientes con tumores que expresen receptores de somatostatina, el tratamiento con análogos puede dar lugar a respuestas bioquímicas y radiológicas marcadas74. El tratamiento con 177Lu-octreotida ha sido eficaz en algunos pacientes80, y es útil en tumores que no captan 131I-MIBG o en combinación con éste ya, que pueden tener efecto sinérgico73. La embolización arterial puede resultar de utilidad en algunas ocasiones. Se ha utilizado empíricamente el tratamiento con bisfosfonatos, con el fin de reducir la resorción ósea, ya que las metástasis suelen ser osteoclásticas. En algunos pacientes puede ser útil la ablación por radiofrecuencia de metástasis hepáticas y óseas81. Si el tumor es agresivo y la calidad de vida se encuentra afectada, se puede considerar el tratamiento con quimioterapia sistémica. La administración de ciclofosfamida, vincristina y dacarbazina en ciclos ha sido beneficiosa, aunque no curativa82. Las crisis adrenérgicas y la hipertensión deben ser controladas con bloqueadores alfa y beta.
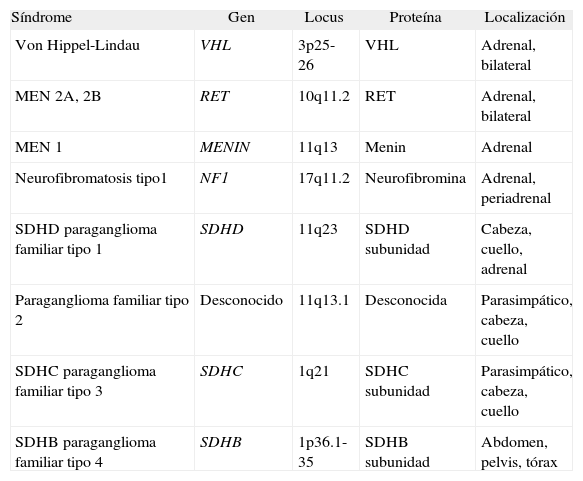
FEOCROMOCITOMA EN SÍNDROMES GENÉTICOSAproximadamente un 15-25% de los pacientes con tumores productores de catecolaminas presentan mutaciones en las líneas germinales en los genes asociados con la enfermedad4,83. Las mutaciones que se identifican con mayor frecuencia en pacientes con feocromocitoma están en los genes asociados con la enfermedad de von Hippel-Lindau (VHL), la neoplasia endocrina múltiple tipo 2 (MEN 2), MEN 1 y la neurofibromatosis tipo 1 (NF1). Estas entidades se asocian con múltiples tumores. En los pacientes con paragangliomas las mutaciones se encuentran en los genes asociados con paraganglioma familiar, VHL y rara vez MEN o NF1. Característicamente suelen aparecer en edades más tempranas. El diagnóstico a menudo se realiza antes de la aparición de síntomas que lo indiquen, como parte de estudios genéticos familiares. En la tabla 6 se exponen los síndromes hereditarios asociados con feocromocitomas y paragangliomas. Todos ellos se heredan con carácter autosómico dominante84. El estudio de familias con las distintas mutaciones ha hecho posible observar las diferencias entre las presentaciones clínicas según la mutación hallada en cada caso. Las correlaciones fenotipo-genotipo describen la asociación entre una mutación específica y un comportamiento clínico particular. Cuando son conocidas, estas correlaciones pueden constituir una guía para el manejo clínico de los pacientes afectados o en riesgo85.
Síndromes hereditarios en relación con feocromocitoma
| Síndrome | Gen | Locus | Proteína | Localización |
| Von Hippel-Lindau | VHL | 3p25-26 | VHL | Adrenal, bilateral |
| MEN 2A, 2B | RET | 10q11.2 | RET | Adrenal, bilateral |
| MEN 1 | MENIN | 11q13 | Menin | Adrenal |
| Neurofibromatosis tipo1 | NF1 | 17q11.2 | Neurofibromina | Adrenal, periadrenal |
| SDHD paraganglioma familiar tipo 1 | SDHD | 11q23 | SDHD subunidad | Cabeza, cuello, adrenal |
| Paraganglioma familiar tipo 2 | Desconocido | 11q13.1 | Desconocida | Parasimpático, cabeza, cuello |
| SDHC paraganglioma familiar tipo 3 | SDHC | 1q21 | SDHC subunidad | Parasimpático, cabeza, cuello |
| SDHB paraganglioma familiar tipo 4 | SDHB | 1p36.1-35 | SDHB subunidad | Abdomen, pelvis, tórax |
MEN: neoplasia endocrina múltiple; SDH: succinato deshidrogenasa.
Tomado de Jiménez C et al86.
Causada por mutaciones del gen VHL, un gen supresor de tumores que codifica una proteína pVHL, que regula la angiogénesis. La alteración del gen VHL afecta a la degradación de los factores inducibles por la hipoxia (HIF). El acúmulo de HIF produce aumento de: factor de crecimiento vascular, factor de crecimiento plaquetario y eritropoyetina. La regulación de genes implicados en el crecimiento celular se pierde, predisponiendo a los portadores a padecer múltiples tumores86. El fenotipo incluye feocromocitoma, frecuentemente bilateral (50%), paraganglioma (10%) en abdomen, tórax y cuello, angiomas retinales, hemangioblastoma cerebeloso, quistes renales y pancreáticos y carcinoma renal. La frecuencia de feocromocitoma en este síndrome es del 10 al 20%. El riesgo de padecer feocromocitoma determina la clasificación de VHL en tipo 1 (no feocromocitoma) y tipo 2 (feocromocitoma) en asociación con otras manifestaciones de la enfermedad. En el tipo 2 se diferencian tres formas según la mutación: A, B y C. En VHL tipo C, el feocromocitoma puede ser la única manifestación, por lo que la presentación puede parecer aparentemente esporádica. Suele producir predominantemente noradrenalina.
Neoplasia endocrina múltipleMEN 1Caracterizado por hiperparatiroidismo primario, tumores pancreáticos y adenomas hipofisarios. Causado por mutaciones del locus MEN1 que codifica una proteína supresora: menina. Puede asociarse con feocromocitoma, aunque la asociación es extremadamente rara y hay pocos casos descritos en la literatura87.
MEN 2Causado por mutaciones activadoras del protooncogón RET, que codifica un receptor de tirosincinasa implicado en la regulación de la proliferación celular y la apoptosis. El MEN 2A incluye feocromocitoma, carcinoma medular de tiroides e hiperparatiroidismo.
El MEN 2B, que es el 5% de todos los MEN 2, incluye feocromocitoma, carcinoma medular de tiroides, neuromas mucosos, ganglioneuromatosis intestinal y hábito marfanoide.
El feocromocitoma aparece aproximadamente en la mitad de los portadores, casi siempre se localiza en las suprarrenales y es bilateral en el 50% de los pacientes. Con frecuencia la aparición de los feocromocitomas es asincrónica, con una separación de hasta 15 años. En general suelen desarrollarse después del carcinoma medular de tiroides, aunque se ha encontrado como la manifestación inicial de este síndrome en mutaciones en el codón 631 que son excepcionales88.
Sin embargo, hay gran variabilidad en la penetrancia del feocromocitoma entre las distintas familias89. La probabilidad de que la enfermedad sea bilateral es mayor que en el caso del feocromocitoma esporádico. El MEN 2 tiene correlaciones genotipo-fenotipo muy específicas; por ejemplo, cualquier mutación en el codón 634 del protooncogén RET resulta en una mayor incidencia de feocromocitoma e hiperparatiroidismo que las mutaciones localizadas en otra región del gen90. Basándose en el seguimiento de 206 portadores con mutaciones en el RET durante 27 años, se concluyó que se debe cribar de feocromocitoma a los pacientes con mutaciones del RET en los codones 918, 634 y 630, anualmente desde los 10 años de edad, y a los portadores de otras mutaciones, desde los 20 años91.
Las características clínicas del MEN y el síndrome VHL son algo diferentes, como han mostrado distintos estudios92: los pacientes con MEN 2 son más sintomáticos, además cursan con concentraciones elevadas de metanefrina, mientras que los pacientes con síndrome VHL presentan concentraciones elevadas de normetanefrina. Los estudios bioquímicos han mostrado diferencias en el almacenamiento y la liberación de aminas asociado con una diferente expresión de las enzimas implicadas en la síntesis de catecolaminas: tirosina hidroxilasa, feniletanolamina N metiltransferasa y aminoácido aromático decarboxilasa93. Los pacientes con VHL tienen menor expresión de estas enzimas, por lo que la síntesis de adrenalina es menor en estos pacientes. Además, también difieren en la expresión de los transportadores encargados de la recaptación de catecolaminas86.
Neurofibromatosis tipo 1En la NF1 la frecuencia del feocromocitoma, que puede ser bilateral (10%) o extraadrenal (6%), varía entre 0,1 y 5,7 según las series94. El cuadro clínico comprende además neurofibromas cutáneos, manchas «café con leche» y hamartomas en el iris. Causado por mutaciones inactivadoras de la neurofibromina, un gen supresor de tumores que codifica una proteína activadora GTP-asa.
PARAGANGLIOMA FAMILIAREn los últimos años ha habido un importante progreso en la identificación de la etiología genética del síndrome del paraganglioma familiar. La mayoría de los casos se deben a mutaciones en tres genes (SDHB, SDHC y SDHD), subunidades codificadoras de la succinato deshidrogenasa o complejo mitocondrial II. La SDH tiene importantes funciones en el ciclo de Krebs y la cadena respiratoria. Los paragangliomas pueden originarse en ganglios simpáticos o parasimpáticos. Los paragangliomas de cabeza y cuello se localizan generalmente en ganglios parasimpáticos y suelen ser negativos a cromafín. Los paragangliomas simpáticos pueden localizarse además en tórax, abdomen, pelvis y vejiga urinaria. La producción de catecolaminas por estos tumores depende de su localización: aproximadamente un 5% de los tumores de cabeza y cuello y un 50% de los abdominales producen hormonas1.
Las mutaciones en SDHD (paraganglioma familiar tipo 1) se han identificado en varias generaciones en algunas familias y se asocian con paragangliomas parasimpáticos de cabeza y cuello95, y con menos frecuencia con feocromocitomas y paragangliomas secretores. Esta mutación predispone al desarrollo de paragangliomas múltiples. La penetrancia depende de si el individuo ha heredado la mutación de la madre o del padre. La enfermedad no se manifiesta cuando la mutación es heredada de la madre, pero la penetrancia es muy elevada cuando se hereda del padre. Este fenómeno se denomina "impresión maternal". Debido a este fenómeno, su presentación puede parecer como esporádica.
El paraganglioma familiar tipo 2 cursa con paraganglioma parasimpático en cabeza y cuello. La mutación no se ha identificado todavía y su frecuencia es muy baja.
Las mutaciones en SDHC que dan lugar al síndrome del paraganglioma familiar tipo 3 se asocian con tumores parasimpáticos de cabeza y cuello96. No se han identificado mutaciones de SDHC en paragangliomas simpáticos o feocromocitomas, por lo que de momento no es necesario considerar esta alteración genética en pacientes con feocromocitomas aparentemente esporádicos79.
Las mutaciones en SDHB causan el síndrome del paraganglioma familiar tipo 4. El síndrome se caracteriza por paragangliomas secretores de catecolaminas a nivel abdominal, torácico y pelviano, así como paragangliomas parasimpáticos. Los pacientes con esta mutación tienen un riesgo más alto de enfermedad maligna. Aunque se ha descrito su asociación a otras neoplasias, como carcinoma de células renales y papilar de tiroides97, en ocasiones puede tener una presentación similar al feocromocitoma esporádico. Al igual que en las mutaciones de SDHC, no se ha demostrado el efecto de la "impresión maternal" observado en los pacientes con mutaciones de SDHD.
Se ha descrito una edad de aparición de la enfermedad más tardía en pacientes con mutaciones en SDHB que las halladas en SDHD98.
Se ha propuesto la clasificación de los feocromocitomas en dos grupos distintos, basándose en las diferencias en las señales de transcripción. Los feocromocitomas con mutaciones en VHL, SDHB y SDHD forman el grupo 1 (cluster 1). Este grupo muestra asociación con la hipoxia y la angiogénesis, y el perfil es consistente con la regulación de la respuesta a la hipoxia dependiente del gen VHL y con la observación de que los factores angiogénicos están sobreexpresados en las mutaciones de SDH79. El grupo 2 (cluster 2) estaría formado por los feocromocitomas asociados a mutaciones del RET o de NF1. Esto concuerda con las diferencias mostradas en el patrón de secreción de adrenalina o noradrenalina que presentan los feocromocitomas asociados con MEN y VHL respectivamente87.
Hay estudios que señalan que la disfunción de la SDH daría lugar a la acumulación de succinato (sustrato de la SDH en el ciclo de Krebs)99. El succinato inhibiría la enzima implicada en la degradación de HIF (prolilhidroxilasa [PHD]), dando lugar a cantidades elevadas de HIF. Otro mecanismo postulado para explicar la tumorigénesis relacionada con la SDH es el aumento en la producción de especies reactivas al oxígeno (ROS), que han mostrado inhibir la actividad PHD, por lo que una disfunción de la SDH conllevaría un aumento en la disponibilidad de HIF79. Estas demostraciones pueden llevar a la identificación de nuevos tratamientos para estas enfermedades.
El patrón expresado por los feocromocitomas en pacientes con mutaciones del RET o de NF1 muestra un aumento en la transcripción de los genes asociados con el metabolismo y la síntesis de ARN.
Feocromocitoma esporádicoSólo en cuatro de los ocho síndromes genéticos descritos, se ha observado una presentación como feocromocitoma aparentemente esporádico (VHL, MEN 2, PGL1 y PGL4). El PGL2 y el PGL3 nunca se han asociado con feocromocitomas y la NF1 puede identificarse con una cuidadosa exploración física. Un estudio europeo reciente ha señalado que un 24% de los pacientes con feocromocitoma aparentemente esporádico tiene una mutación en las líneas germinales: el 11% en VHL, el 4,8% en RET, el 4,8% en SDHB y el 4,6% en SDHD. Estos hallazgos llevaron a la recomendación de que el estudio genético debía considerarse en todos los pacientes con paraganglioma o feocromocitoma100. La revisión de todos los estudios publicados con datos sobre mutaciones en las líneas germinales indica a primera vista que un 20% de los pacientes diagnosticados de feocromocitoma esporádico son portadores de alguna mutación en alguno de los genes conocidos como causantes de feocromocitomas hereditarios. Excluyendo a los pacientes con enfermedad bilateral o multicéntrica, la cifra se reduce al 17,5% (el 5,04% en VHL, el 1,55% en RET, el 3,72% en SDHB y el 6,38% en SDHD)79. La existencia de mutaciones espontáneas, baja penetrancia, protección materna e interacciones entre genes o con el ambiente puede explicar en parte este fenómeno4. Además se han observado diferencias importantes entre distintas poblaciones101,102.
Debe confirmarse la existencia de una mutación en un paciente afecto antes de ofrecer estudio genético a sus familiares asintomáticos103. Es importante realizar una exploración clínica del paciente que permita hallazgos clínicos que indiquen enfermedad genética. Así, el clínico debe considerar diversos factores como: la localización del tumor, producción hormonal, malignidad, multicentricidad e historia familiar antes de decidir que mutación debe estudiarse en primer lugar.
El grupo europeo para el estudio de tumores adrenales y el grupo de trabajo sobre feocromocitoma recomiendan el estudio genético de todos los pacientes con feocromocitopma o paraganglioma104. Las indicaciones del estudio genético se exponen en la tabla 7.
Consideraciones sobre el estudio genético
| Orden a seguir | |
| Estudio genético | |
| Feocromocitoma unilateral en paciente < 20 años* | VHL > RET > SDHB = SDHD |
| Feocromocitoma bilateral* | VHL > RET > SDHB = SDHD |
| Feocromocitoma unilateral en paciente > 20 años, sin hallazgos clínicos o historia familiar sugestiva* | SDHB + VHL > SDHD > RET |
| Paraganglioma cabeza y cuello | SDHD > SDHC > SDHB |
| Paraganglioma abdominal | SDHB > SDHD > VHL |
| Feocromocitoma unilateral en paciente > 50 años, sin hallazgos clínicos o historia familiar sugestiva | Estudio genético opcional |
El feocromocitoma es una causa excepcional de hipertensión en el embarazo y suele cursar con un cuadro clínico similar que la población general. Sin embargo, debido a la compresión del útero grávido, las pacientes pueden presentar crisis hipertensivas paroxísticas en la posición supina pero mantener cifras normales de presión arterial en otras. Si hay hipertensión y proteinuria, es difícil distinguirlo de la preeclampsia, situación en la que se ha observado además un aumento de la excreción de noradrenalina. El diagnóstico se basa en la determinación de metanefrinas y catecolaminas fraccionadas en orina, así como metanefrinas plasmáticas. En cuanto al diagnóstico de localización se refiere, la técnica de elección es la RM105.
Dado que hay pocos casos descritos, no existe un tratamiento óptimo, pero se recomienda iniciar la preparación prequirúrgica con fenoxibenzamina106. La fenoxibenzamina cruza la placenta y puede producir depresión perinatal e hipotensión transitoria. El uso de bloqueadores beta no es aconsejable, ya que puede producir retraso de crecimiento intrauterino. También conviene evitar los IECAS y los ARA-II. El tratamiento definitivo debe ser quirúrgico y la cirugía laparoscópica es la técnica recomendada107. La cirugía debe realizarse en el segundo trimestre, aunque la indicación no está realmente clara108. En las pacientes diagnosticadas en el tercer trimestre se debe tratar con bloqueadores alfa y antagonistas del calcio y realizar una cesárea y la resección del tumor cuando el embarazo esté prácticamente a término. La cesárea es preferible a la vía vaginal, ya que parece conllevar menor riesgo de mortalidad materna109.
PRONÓSTICOLa cirugía del feocromocitoma no siempre conduce a la curación de los pacientes, incluso con tumores benignos. Se han descrito recidivas en un 16% de los pacientes en algunas series. La recidiva es más frecuente en pacientes jóvenes, enfermedad familiar, afección bilateral o extraadrenal, y tumores localizados en la suprarrenal derecha (el 62 frente al 7%). Así pues, el seguimiento clínico y bioquímico de estos pacientes debe ser indefinido, realizando controles anuales y bianuales a los sujetos con tumores suprarrenales izquierdos110. En la enfermedad maligna la tasa de supervivencia a los 5 años es menor del 50%.
