




El estudio genético del protooncogén RET permite un diagnóstico precoz del síndrome de neoplasia endocrina múltiple tipo 2 y establece una correlación entre el genotipo y las manifestaciones clínicas. El objetivo del presente trabajo es demostrar los beneficios del diagnóstico precoz por estudio genético seguido de tratamiento temprano en la curación del carcinoma medular de tiroides (CMT) frente al diagnóstico más tardío con la calcitonina sérica.
Pacientes y métodoEstudio descriptivo retrospectivo de 8 miembros de una familia con MEN2A por mutación C634Y. Se realizó despistaje con calcitonina sérica hasta 1999 y estudio genético de RET posteriormente. A los portadores se les realizó tiroidectomía total y determinaciones periódicas de calcitonina, metanefrinas urinarias, calcio, fósforo y pruebas de imagen a nivel cervical y abdominal.
ResultadosLos 5 pacientes diagnosticados por despistaje familiar con calcitonina presentan en la actualidad cifras de calcitonina elevadas. Los 3 diagnosticados por estudio genético (un adulto y dos niños) se encuentran libres de enfermedad. En los niños se monitorizó la calcitonina y se les intervino cuando esta comenzó a elevarse, a los 6 y 10 años respectivamente, hallándose hiperplasia nodular de células C en ambos. De los 8 afectos 3 presentaron feocromocitomas, bilaterales y asincrónicos, la mitad con metanefrinas urinarias normales y dos simultáneos al CMT. Ningún paciente presentó alteraciones bioquímicas sugestivas de hiperparatiroidismo aunque en uno se descubrieron adenomas paratiroideos múltiples durante la cirugía tiroidea.
ConclusionesEl estudio genético de RET ha conseguido el diagnóstico y tratamiento precoces y por tanto la curación del CMT en nuestros pacientes, orientándonos sobre el momento y tipo de cirugía adecuados y permitiendo correlacionar fenotipo-genotipo, ejemplificando cómo una alteración genética se asocia a patología que podemos prever y manejar mejorando el pronóstico de nuestros pacientes.
Genetic testing of RET proto-oncogen allows an early diagnosis of Multiple Endocrine Neoplasia syndrome type 2 and establish a correlation between genotype and clinical manifestations. The purpose of this study was to demonstrate the benefits of an early diagnosis with genetic testing followed by prompt surgery on the cure of MTC versus a later diagnosis with serum calcitonin.
Patients and methodRetrospective descriptive study of 8 members of a MEN 2A family by C634Y mutation. We performed serum calcitonin screening until 1999 and subsequently RET genetic testing was obtained. Carriers underwent total thyroidectomy and periodic determination of calcitonin, urinary metanephrines, calcium, phosphorus and neck and abdominal imaging techniques.
ResultsFive patients were diagnosed by calcitonin familial screening and all of them have high calcitonin by now. Three patients were diagnosed by genetic testing (an adult and two children) and they are free of disease. Calcitonin was closely monitored in children and they underwent surgery when it started to raise, at 6 and 10 years old respectively, finding nodular C-cell hyperplasia in both. Of 8 carriers 3 developed pheochromocytomas, bilateral and asynchronous, one-half had normal urinary metanephrines and two of them were simultaneous with MTC. No patient had biochemical data suggesting hyperparathyroidism although in one patient multiple parathyroid adenomas were found at thyroidectomy.
ConclusionsRET genetic analysis has achieved an early diagnosis and treatment with no development of MTC in our patients, adjusting the time and type of surgery and allowing a genotype-phenotype correlation. It demonstrates how a genetic alteration is associated with a pathology that we can prevent and manage improving the prognosis of our patients.
El síndrome de neoplasia endocrina múltiple tipo 2 (MEN2A) fue descrito en 1961 por Sipple, quien propuso la asociación entre el feocromocitoma bilateral y el carcinoma de tiroides1. Posteriormente Steiner acuñó el término MEN2 y observó que el fenotipo era más amplio e incluía: carcinoma medular de tiroides (CMT), feocromocitoma e hiperparatiroidismo. En el MEN2A el CMT es el elemento prínceps, ya que se presenta en más del 95% de los pacientes, mientras que el feocromocitoma ocurre en el 50% y el hiperparatiroidismo entre el 10 y el 30%2–4. Además, el periodo de latencia hasta la aparición completa del síndrome es amplio, pudiendo tardar varias décadas. Existen 2 variantes, una con enfermedad de Hirschprung y otra con amiloidosis cutánea. El MEN2B se caracteriza por CMT de especial agresividad, feocromocitoma, ganglioneuromas y hábito marfanoide. EL CMT familiar (CMTF) es una variante incompleta en la que solo se desarrolla CMT aunque ocasionalmente se asocia a enfermedad de Hirschprung o a liquen amiloideo cutáneo.
El MEN2 es un trastorno genético, con herencia autosómica dominante y penetrancia de casi el 100%, causado por mutaciones missense (cambio de un aminoácido por otro) en la línea germinal del protooncogén RET (REarregement during Transfection). El gen RET se sitúa en el cromosoma 10 (10q11.2) y está constituido por 21 exones. Codifica un receptor tirosín cinasa que se expresa en las células derivadas de la cresta neural e interviene en procesos de crecimiento y diferenciación celular. Sus mutaciones afectan fundamentalmente a 4 tipos de tejidos, todos derivados de la cresta neural: células C tiroideas, paratiroides, células cromafines de la médula adrenal y plexo autonómico entérico5. En el CMT el desarrollo del tumor se produce por la activación constitutiva del receptor, que en el MEN2A conduce a una proliferación celular acelerada6. La sensibilidad a la activación de RET es distinta en cada tejido, por lo que el feocromocitoma y el hiperparatiroidismo solo ocurren en determinadas mutaciones, especialmente las del codón 634.
Se cree que la incidencia de mutación de RET es de 1 portador por cada 500.000 habitantes/año7. La prevalencia estimada de MEN2 es de 1 por cada 30.000 habitantes, siendo más del 80% de estos MEN2A8. En general la edad de presentación del CMT y la frecuencia de afectación paratiroidea y de la médula adrenal no dependen del tipo de aminoácido que sustituye al original, sino del codón donde se produce este cambio; si bien algunas mutaciones específicas sí se relacionan con el curso de la enfermedad9. Se postula que el riesgo de progresión depende del potencial de transformación que posee la mutación en cada individuo10. Hasta ahora se han descrito diferentes mutaciones en RET asociadas a MEN2A; las del codón 634 son las más frecuentes, englobando el 80% de las mutaciones de RET en línea germinal. Esta mutación condiciona un fenotipo clínico abigarrado, pudiendo expresarse como CMT familiar (CMTF) o MEN2A. Los portadores desarrollan CMT en virtualmente el 100% de los casos, de aparición precoz y con un alto índice de metástasis, persistencia o recurrencia11, por lo que se cataloga como mutación de alto riesgo12. Se relaciona estrechamente con el feocromocitoma4,11 y esporádicamente con el hiperparatiroidismo. La mutación en el codón 634 se asocia con liquen amiloideo cutáneo y no tiene relación causal con la enfermedad de Hirschprung.
En este sentido se sabe que existe una correlación genotipo-fenotipo, es decir, conociendo el codón mutado podemos prever a qué edad se desarrollará el CMT. Esto ha conducido a elaborar grupos de riesgo y recomendaciones específicas en cuanto al momento y tipo de cirugía adecuados, basándose en el paciente más joven diagnosticado de CMT, la edad mínima de presentación de metástasis ganglionares y la edad media de presentación del CMT en cada familia con una mutación determinada4. La tiroidectomía profiláctica en los portadores de la mutación ha cambiado la historia natural de la enfermedad, constituyendo el caso más representativo de prevención primaria de un cáncer genético13.
No se conoce con exactitud por qué dentro de una familia con la misma mutación unos desarrollan feocromocitoma o hiperparatiroidismo y otros no14, aunque se plantea la contribución de factores ambientales y genéticos como los polimorfismos de RET. Dichos factores, junto con la aparición de mutaciones somáticas, podrían justificar el fenómeno de anticipación genética según el cual el fenotipo es más agresivo conforme pasan las generaciones. De ahí la importancia de seguir a lo largo de la vida a los afectos con mutación RET.
Nos proponemos realizar un estudio descriptivo y evolutivo de una familia con MEN2A por mutación C634Y en el protooncogén RET seguida durante más de 20 años.
Sujetos y métodoSe estudiaron 20 miembros pertenecientes a 3 generaciones de una familia con MEN2A por mutación C634Y. A partir del caso índice (paciente 1; tabla 1) que se diagnosticó de CMT en 1989 se realizó estudio a familiares de primer grado, con calcitonina sérica antes de 1999 y posteriormente con estudio genético del protooncogén RET, que también se hizo a los hijos de los portadores. Una rama rechazó seguimiento (fig. 1). La mutación pudo ser transmitida por línea paterna, ya que la madre del caso índice falleció a los 87 años mientras que el padre lo hizo antes de los 40 por causas no conocidas.
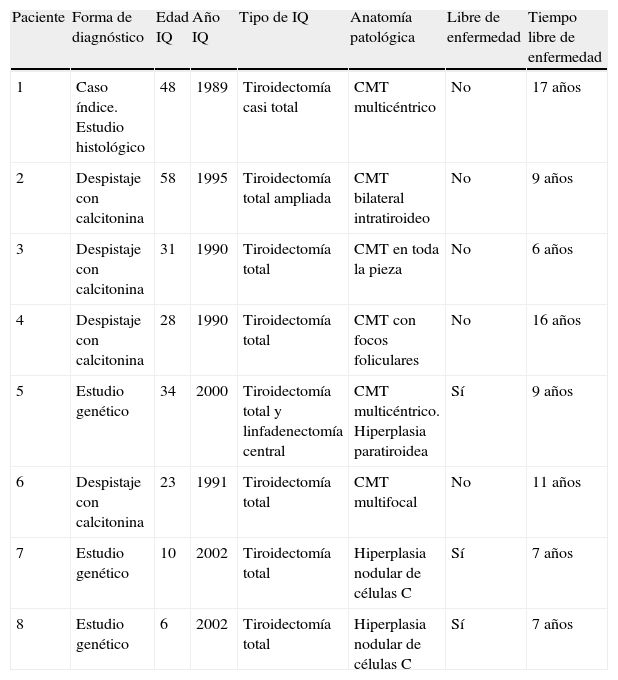
Características del carcinoma medular de tiroides en familia con MEN 2A
| Paciente | Forma de diagnóstico | Edad IQ | Año IQ | Tipo de IQ | Anatomía patológica | Libre de enfermedad | Tiempo libre de enfermedad |
| 1 | Caso índice. Estudio histológico | 48 | 1989 | Tiroidectomía casi total | CMT multicéntrico | No | 17 años |
| 2 | Despistaje con calcitonina | 58 | 1995 | Tiroidectomía total ampliada | CMT bilateral intratiroideo | No | 9 años |
| 3 | Despistaje con calcitonina | 31 | 1990 | Tiroidectomía total | CMT en toda la pieza | No | 6 años |
| 4 | Despistaje con calcitonina | 28 | 1990 | Tiroidectomía total | CMT con focos foliculares | No | 16 años |
| 5 | Estudio genético | 34 | 2000 | Tiroidectomía total y linfadenectomía central | CMT multicéntrico. Hiperplasia paratiroidea | Sí | 9 años |
| 6 | Despistaje con calcitonina | 23 | 1991 | Tiroidectomía total | CMT multifocal | No | 11 años |
| 7 | Estudio genético | 10 | 2002 | Tiroidectomía total | Hiperplasia nodular de células C | Sí | 7 años |
| 8 | Estudio genético | 6 | 2002 | Tiroidectomía total | Hiperplasia nodular de células C | Sí | 7 años |
IQ: intervención quirúrgica.

El seguimiento del CMT se ha llevado a cabo mediante cuantificación periódica de niveles séricos de calcitonina, ecografía cervical y realización de octreoscan a partir del año 2000 cuando existía aumento de calcitoninemia sin que se demostraran restos tiroideos por técnicas de imagen (ecografía cervical y TAC cervicotorácica).
En todos los portadores se cuantificaron metanefrinas urinarias anualmente mediante HPLC y se solicitaron técnicas de imagen abdominal: RMN cada 3 años en los adultos y ecografía anual en los niños. Para la detección de hiperparatiroidismo se monitorizaron anualmente los niveles de calcio y fósforo, que hasta la fecha han sido normales en todos los pacientes.
ResultadosCarcinoma medular de tiroidesDe los 8 pacientes 3 fueron diagnosticados por estudio genético, dos a los 2 y 3 años (pacientes 7 y 8) y uno en la edad adulta (paciente 5). En los niños se decidió determinación anual de calcitonina y se programó la cirugía cuando esta alcanzó el límite superior de la normalidad, que fue a los 10 años en el paciente 7 y a los 6 en el paciente 8. La técnica quirúrgica fue tiroidectomía total y la anatomía patológica fue hiperplasia nodular de células C en ambos. Siete años después se encuentran libres de enfermedad y sin secuelas derivadas de la intervención. El paciente 5 se diagnosticó a los 34 años, se intervino mediante tiroidectomía total con linfadenectomía central detectándose CMT multicéntrico con ausencia de afectación ganglionar e igualmente no tiene datos de enfermedad en el momento actual.
El caso índice y los 4 familiares diagnosticados por despistaje bioquímico con calcitonina no se han curado, según indican los valores elevados de calcitonina, pese a que varios mantuvieron remisiones prolongadas (tabla 1). En ninguno de ellos se realizó linfadenectomía cervical, por lo que desconocemos si existían metástasis ganglionares al diagnóstico.
El tiempo medio de remisión de la enfermedad, entendido como calcitonina sérica basal indetectable o en el rango de la normalidad, fue de 11,8 años.
La anatomía patológica mostró distintas variedades de CMT, en todos los casos multicéntrico y bilateral, en el paciente 5 además acompañado de hiperplasia paratiroidea.
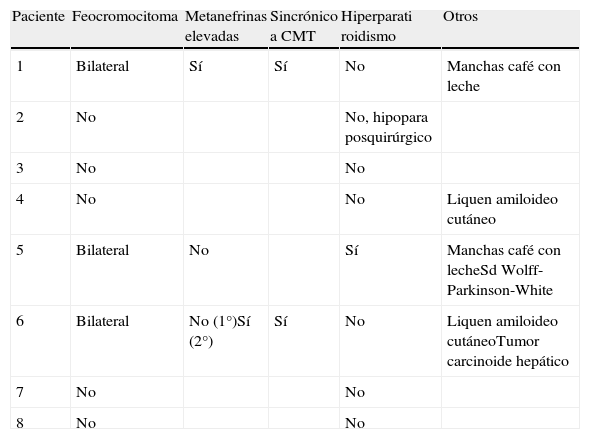
FeocromocitomaTres de los 8 pacientes presentaron feocromocitomas, bilaterales y asincrónicos. Tres fueron detectados por metanefrinas urinarias elevadas y tres por técnicas de imagen. El caso índice (paciente 1) fue el único tiroidectomizado sin estudio previo de feocromocitoma. En el paciente 5 las metanefrinas urinarias antes de la cirugía tiroidea resultaron normales, demostrándose posteriormente por técnicas de imagen la presencia de una masa adrenal que resultó ser un feocromocitoma.
HiperparatiroidismoSolo el paciente 5 presentó hiperplasia de paratiroides, no diagnosticada antes de la cirugía tiroidea, ya que tenía niveles de calcio y fósforo normales. La exéresis de las paratiroides macroscópicamente aumentadas de tamaño le ha condicionado un hipoparatiroidismo grave con altos requerimientos de calcio y vitamina D.
OtrosRespecto a otros hallazgos asociados a MEN2, se encontraron 2 sujetos con liquen amiloideo cutáneo. Ninguno padece la enfermedad de Hirschprung. Otros hallazgos no relacionados con MEN2 se muestran en la tabla 2.
Neoplasias asociadas a MEN2A y otros hallazgos clínicos
| Paciente | Feocromocitoma | Metanefrinas elevadas | Sincrónico a CMT | Hiperparati roidismo | Otros |
| 1 | Bilateral | Sí | Sí | No | Manchas café con leche |
| 2 | No | No, hipopara posquirúrgico | |||
| 3 | No | No | |||
| 4 | No | No | Liquen amiloideo cutáneo | ||
| 5 | Bilateral | No | Sí | Manchas café con lecheSd Wolff-Parkinson-White | |
| 6 | Bilateral | No (1°)Sí (2°) | Sí | No | Liquen amiloideo cutáneoTumor carcinoide hepático |
| 7 | No | No | |||
| 8 | No | No |
El MEN2A o síndrome de neoplasia endocrina múltiple tipo 2A es debido a mutaciones en el protooncogén RET. Su caracterización ha supuesto un cambio absoluto en el manejo gracias al diagnóstico y tratamiento tempranos que han condicionado satisfactoriamente su pronóstico. El estudio genético ha permitido no solo un diagnóstico precoz del CMT, sino un manejo dirigido a los portadores previo al desarrollo de la enfermedad, a diferencia del estudio familiar con calcitonina sérica, realizado a todos los familiares de primer grado a lo largo de la vida, y cuya elevación ya reflejaba la presencia de tumor en nuestro estudio. El tratamiento precoz en el CMT constituye el paradigma de la prevención primaria del cáncer hereditario en seres humanos13. Por otra parte, la existencia de una mutación determinada se asocia a una presentación clínica característica lo que permite hacer una correlación genotipo-fenotipo fundamental en el abordaje del paciente.
De las múltiples mutaciones descritas la del codón 634 es la más frecuente y, obviamente, la más conocida. Esta mutación condiciona una alteración del receptor tirosín cinasa codificado por RET, confiriéndole una ganancia de función por dimerización del receptor independiente de ligando y activación constitutiva del mismo. Esta mutación se ha asociado con todas las manifestaciones descritas en el MEN2A.
En nuestra serie solo los tratados precozmente gracias al estudio genético se encuentran libres de enfermedad, mientras que el resto tiene datos de persistencia o recurrencia. Podría plantearse la peor evolución del grupo diagnosticado bioquímicamente por la ausencia de linfadenectomía central profiláctica como aconsejan las guías actuales12 o porque el diagnóstico con calcitonina, más tardío, les confiriera peor pronóstico al hallarse en un estadio más avanzado; esto no se confirmó histológicamente por la no realización de linfadenectomía y biopsia de ganglios laterocervicales y mediastínicos, ya que se intervinieron años antes de que se elaboraran las guías actuales que recomiendan estas prácticas.
Carcinoma medular de tiroidesEl CMT en la mutación 634 se presenta de forma precoz, con agresividad y gran capacidad metastásica, lo que implica altas tasas de persistencia o recurrencia tras la cirugía. En nuestros pacientes hemos encontrado intervalos libres de enfermedad de hasta 17 años seguidos de recurrencias. El desarrollo del CMT se inicia con la hiperplasia de células C, un estadio pretumoral, que evoluciona de manera muy dispar a lo largo del tiempo, formando focos bilaterales y multicéntricos. Es la primera neoplasia en desarrollarse y es la causa de muerte más frecuente en pacientes con MEN2. En nuestra serie no hubo ningún fallecimiento pese a que la literatura refleja tasas de supervivencia a 10 años de entre el 100 y el 71% en los estadios I-III del CMT, que disminuye al 21% en el estadio IV15 aunque no existen datos concretos en MEN2A.
La tiroidectomía profiláctica es la práctica habitual en los portadores de la enfermedad. Si bien la indicación sería antes de los 5 años de edad por ser una mutación de alto riesgo12 (tabla 3), en nuestro caso decidimos mantener un seguimiento estrecho y programar la cirugía ante discretas elevaciones de calcitonina, aun dentro del rango de normalidad. Esta decisión se consensuó con los padres ya que eran reacios a una cirugía en la primera infancia. Hay que tener en cuenta que la edad de presentación en esta familia no había sido especialmente temprana y su evolución puede considerarse bastante benigna, con un 100% de supervivencia hasta la fecha. Siendo conocedores del estudio genético confiaban plenamente en el valor diagnóstico de la calcitonina. Por todo esto la tiroidectomía profiláctica se realizó a los 6 y 10 años de edad en los pacientes 7 y 8 con resultado de hiperplasia de células C.
Estratificación en grupos de riesgo según la 7th International MEN Meeting 1999
| Grupo de riesgo | Indicación del momento y tipo de cirugía |
| Grupo 1: bajo riesgo. Codones 609, 768, 790, 791, 804, 891 | Tiroidectomía total entre los 5 y 10 años. O determinación anual de calcitonina y programar la cirugía cuando se eleve |
| Grupo 2: alto riesgo. Codones 611, 618, 620, 634 | Tiroidectomía total con o sin linfadenectomía central antes de los 5 años |
| Grupo 3: máximo riesgo. MEN2B. Codones 883, 918 y 922 | Tiroidectomía total con linfadenectomía central en los 6 primeros meses de vida |
La correlación genotipo-fenotipo tiene ciertas limitaciones. Se sabe que mutaciones en determinados codones pueden originar diferentes expresiones clínicas16, en los exones 10 y 11 (dominio extracelular) se asocian con MEN2A o CMTF. Algunas mutaciones se asocian con el desarrollo de CMT y otros tumores endocrinos cuando se presentan en homocigosis o se asocian a mutaciones somáticas17. La edad de presentación del CMT puede variar si se pierde el alelo normal de RET o se duplica el mutado17. Asimismo, aunque la correlación genotipo-fenotipo es útil para el CMT no predice con precisión el desarrollo de hiperparatiroidismo o feocromocitoma ni la edad de aparición de los mismos.
FeocromocitomaLos feocromocitomas en el MEN2A con mutación 634 son más frecuentes que en otras mutaciones. Característicamente son bilaterales, no malignizan y no se encuentran en localizaciones extraadrenales. El feocromocitoma es la primera manifestación de MEN en el 25% de los casos, es concomitante con el CMT en el 35% y posterior a este en el 40%18–20. En nuestra serie ningún paciente fue diagnosticado de feocromocitoma antes que de CMT. En mutaciones del codón 634 se han descubierto en niños de 5 a 10 años, por lo que el cribado debe comenzarse antes de realizar la tiroidectomía o cualquier otra cirugía y continuarse cada año. Para el despistaje se recomienda la realización de catecolaminas fraccionadas y metanefrinas en orina de 24 h, si bien algunos recomiendan las metanefrinas séricas por su mayor sensibilidad, no confirmada en algunos estudios21. En nuestra serie solo la mitad presentaron metanefrinas elevadas en orina. Las guías recomiendan la realización de pruebas de imagen en el momento del diagnóstico bioquímico y cada 3 a 5 años a partir de los 15 años10.
HiperparatiroidismoEl hiperparatiroidismo es el hallazgo menos frecuente del MEN2A. Se asocia con mutaciones del codón 634 por lo que se recomienda determinación anual de calcio sérico y PTH, ya que la mayoría de las veces es asintomático. Durante la cirugía tiroidea de un paciente normocalcémico con MEN2A se pueden encontrar una o más paratiroides aumentadas de tamaño como ocurrió con el paciente 5. La resección profiláctica puede condicionar hipocalcemia permanente y es cuestionable22.
Liquen amiloideoEs una lesión cutánea pruriginosa, que se localiza habitualmente en la parte superior de la espalda. No todos los pacientes con liquen amiloideo tienen mutaciones en RET pero cuando se asocia a CMT habitualmente se trata de mutaciones en el codón 634, exceptuando un caso con mutación en el 80423. Se cree que puede estar relacionado con una anomalía sensitiva en los dermatomas C6-T6 que conllevaría prurito neurológico y depósito de amiloide como consecuencia del rascado repetido24. En nuestra familia hay 2 casos, ambos detectados varios años después del diagnóstico de CMT aunque también se han descrito antes que este.
ConclusionesEl MEN 2A es un síndrome genético con clínica abigarrada que condiciona importante morbilidad. El estudio genético del protooncogén RET ha conseguido el diagnóstico y tratamiento precoces del CMT en nuestros pacientes, siendo determinante en mantener en remisión tras la cirugía a los que ya habían desarrollado. Además, nos ha orientado sobre el momento y tipo de cirugía adecuados permitiendo correlacionar fenotipo-genotipo, siendo la mutación 634 un ejemplo de cómo una alteración genética se asocia a patología que podemos prever y manejar mejorando de forma significativa el pronóstico de estos pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
