


Introducción
La amiloidosis comprende un grupo de enfermedades sistémicas y localizadas, en las que se produce el depósito patológico extracelular de proteínas fibrilares en forma de hoja plegada en distintas zonas del organismo. Se conocen diversas formas y clasificaciones de amiloidosis. Todas las formas comparten la glucoproteína amiloidea P (SAP) y difieren en la proteína fibrilar. En la amiloidosis primaria (AL) aparecen porciones variables de cadenas ligeras de inmunoglobulinas como resultado de una discrasia de células plasmáticas1. En ella el número de clones de células plasmáticas en la médula ósea es pequeño, y es frecuente la detección por inmunoelectroforesis de inmunoglobulinas amiloidogénicas en suero u orina2. Existe un 26 % de amiloidosis AL que se asocia a mieloma múltiple y que por tanto no se considera como idiopática3. El objetivo de nuestro estudio fue conocer la historia natural, la afectación de los distintos órganos y la evolución clínica de los pacientes diagnosticados de amiloidosis primaria en nuestro servicio, entre mayo de 1998 y abril del 2005
Pacientes y método
Estudiamos retrospectivamente los casos de amiloidosis primaria diagnosticados en el período señalado. El diagnóstico de amiloidosis primaria se basó en la presencia de depósitos rojo congo en las biopsias realizadas: renal, gástrica o de médula ósea. Fue necesaria la realización de inmunohistoquimia con resultado positivo para cadenas monoclonales k y l y negativo para anticuerpos antiproteína AA. Los pacientes con una presencia mayor del 10 % de células plasmáticas en la médula ósea, lesiones osteolíticas o un componente M sérico o urinario muy elevado se desestimaron por considerarse asociados a mieloma múltiple4.
De las historias clínicas se recogieron las características generales, los antecedentes personales, los hallazgos clínicos y de laboratorio, así como el tratamiento y la supervivencia de los pacientes. Los resultados se expresaron como media ± desviación estándar5. El análisis estadístico se realizó mediante el programa R Sigma Babel y se utilizaron las curvas de supervivencia de Kaplan Meier.
A continuación realizamos una breve descripción de las manifestaciones de la enfermedad en cada uno de nuestros pacientes.
Caso 1
Varón de 78 años con hipotensión sintomática y disnea de pequeños esfuerzos. Refería crisis de dolor abdominal intenso, y en la exploración se apreció anasarca. En las exploraciones analíticas destacaba un pico monoclonal sérico de IgM de 2.165 mg/dl, albúmina de 1,7 g/dl, creatinina de 1,1 g/dl, gammaglutamiltransferasa (GGT) de 195, bilirrubina de 4,77 mg/dl y fosfatasa alcalina de 770 U/l, junto a proteinuria de 7 g/día. El paciente presentaba proteinuria de Bence-Jones débilmente positiva para cadenas l. En la biopsia de médula ósea se encontraron depósitos rojo congo positivos en la pared vascular. En la biopsia renal destaca la expansión del espacio mesangiocapilar por la presencia de material amorfo rojo congo positivo. El paciente fue tratado con corticoides y melfalán, sin respuesta. Desarrolló fracaso multiorgánico y falleció a los 2 meses.
Caso 2
Varón de 67 años con antecedentes de insuficiencia cardíaca congestiva e hipertensión arterial. Presentaba una foria descompensada y parálisis del IV y VII pares craneales. Acudió con creatinina de 6,26 y urea de 56 mg/dl, junto con GGT de 203 y fosfatasa alcalina de 352 U/l. No presentó picos monoclonales,
ni proteinuria. Durante el ingreso destaca la clínica de ortopnea y dolor abdominal intenso. En la biopsia renal aparecen amplios depósitos rojo congo positivos que borran la estructura de arterias y arteriolas. Además hay una atrofia del 20 % del parénquima, con los túbulos conservados. Se le administró tratamiento con melfalán y prednisona y se le incluyó en programa de hemodiálisis. Al año desarrolló una neumonía y falleció por caquexia, a los 18 meses del diagnóstico.
Caso 3
Varón de 71 años con antecedentes de enolismo y cirrosis hepática, estadio C de Child-Pugh. Fue diagnosticado de macroglobulinemia de Waldenström y desarrolló una grave polineuropatía sensitivomotora y un estado de anasarca que motivó su ingreso. Destacamos la creatinina de 2,8 y la bilirrubina de 9,77 mg/dl. La médula ósea se encontró hipocelular, con un infiltrado de células linfoides pequeñas y maduras. La biopsia renal demostró depósitos rojo congo positivos en todos los glomérulos. Tras permanecer un solo mes en hemodiálisis, falleció por una situación de caquexia progresiva.
Caso 4
Varón de 61 años con un cuadro de deterioro progresivo del estado general y cefaleas acompañadas de alteración de la visión. Presentó epigastralgias, vómitos y mareos. Destacaba la creatinina de 2,79 mg/dl, la fosfatasa alcalina de 778 U/l y la proteinuria de 12 g/día, con Bence-Jones positivo k. La médula ósea fue hipocelular, sin infiltración de células plasmáticas atípicas, y en la biopsia gástrica aparecieron depósitos rojo congo positivos. Se instauró tratamiento con melfalán y prednisona, con mala evolución clínica, y falleció en el contexto de una septicemia por bacilos gramnegativos a los 2 meses de su ingreso.
Caso 5
Mujer de 76 años con síndrome metabólico e hipotiroidismo subclínico que se remite para estudio de proteinuria de 5,8 g/día con función renal normal. Presenta pérdida de 6 kg en 2 meses, una polineuropatía sensitiva e hipotensión arterial. Además desarrolla una bacteriemia por Escherichia coli. En la biopsia de la médula ósea existen depósitos rojo congo en la pared de los vasos. La supervivencia fue de 4 meses.
Caso 6
Mujer de 67 años seguida en consultas externas por síndrome nefrótico con función renal preservada. En la biopsia renal sólo se evidenciaba un mínimo depósito rojo congo positivo en la pared de un vaso. Al año acude por presentar disnea a moderados esfuerzos y epigastralgia. Entonces presentaba deterioro de función renal, con creatinina de 5,17 mg/dl. En la ecocardiografía se observa hipertrofia concéntrica del ventrículo izquierdo y disfunción diastólica. Se realiza gastroscopia y biopsia, con la presencia en ésta de depósitos congófilos. La posterior punción de médula ósea recoge un 2 % de células plasmáticas. La paciente no recibió tratamiento quimioterápico y actualmente lleva 12 meses en programa de hemodiálisis. Como complicación presenta repetidos episodios de hemorragia digestiva aguda.
Caso 7
Varón de 84 años con antecedentes de diabetes mellitus tipo 2 e hipertrofia de próstata que es remitido para estudio de síndrome nefrótico con proteinuria de 7,86 g/día. Durante el estudio se observa un pico IgG plasmático. Desarrolla progresiva insuficiencia renal crónica, hasta grado 4. En la biopsia renal se aprecian depósitos rojo congo positivos. La supervivencia actual es de 9 meses.
Caso 8
Mujer de 79 años con diabetes mellitus tipo 2, cardiopatía isquémica, enfermedad del seno con taquicardias y bradicardias sintomáticas, portadora de marcapasos definitivo y un ictus de la arteria cerebral anterior izquierda trombolisado. Acude por anasarca junto a cuadro constitucional, con pérdida de 9 kg en los 2 últimos meses. Presenta síndrome nefrótico y síncopes posturales. En la biopsia renal se apreció expansión mesangionodular acompañada de depósitos algodonosos rojo congo positivos. La paciente actualmente presenta una supervivencia de 17 meses, con buena calidad de vida.
Caso 9
Mujer de 67 años con síndrome metabólico y artrosis generalizada. Cabe destacar una clínica de síncopes ortostáticos de repetición. Al diagnóstico la creatinina era de 3,16; la urea, de 129 mg/dl; la fosfatasa alcalina, de 409 U/l, y la velocidad de sedimentación globular (VSG), de 120 mm/h. La proteinuria era de 2,32 g/día, con Bence-Jones positiva. En la punción de médula ósea se cuantificó un 9 % de células plasmáticas atípicas. En la biopsia renal destaca afectación difusa glomerular con un abundante material rojo congo e inmunohistoquimia k y l positivas. La paciente fue tratada con varios regímenes de inmunosupresores, como VAD (vincristina, doxorrubicina y dexametasona) y otros. Posteriormente se le realizó un trasplante de médula ósea autólogo. Permaneció 2 meses en hemodiálisis, con recuperación posterior de función renal y supervivencia de 22 meses, hasta la actualidad.
Resultados
De los 9 pacientes recogidos en el estudio, 5 eran varones y 4 mujeres. La edad al diagnóstico fue de 72,31 ± 19,11 años (intervalo de 61 a 84 años).
El síndrome constitucional fue un hallazgo habitual presente en 5 pacientes. Se calculó que la pérdida de peso presentaba una mediana de 6 kg en 2 meses. Aparecieron neuropatías autonómicas6, tales como hipotensiones ortostáticas y síncopes posturales. Fue reiterada la aparición de molestias digestivas sin localización clara4 e ictericia franca2. Dos pacientes presentaron hepatomegalia, y uno, macroglosia.
En la ecocardiografía se demostró en 5 casos hipertrofia del ventrículo izquierdo y disfunción diastólica. Además, uno de los pacientes también presentó disfunción sistólica. El 66,6 % presentó disnea al diagnóstico. La conducción del estímulo eléctrico cardíaco también mostró alteraciones poco específicas. Dos pacientes presentaron bloqueo de rama derecha, 2 hemibloqueo anterior y 2 signos de isquemia de cara inferior junto a bajos voltajes.
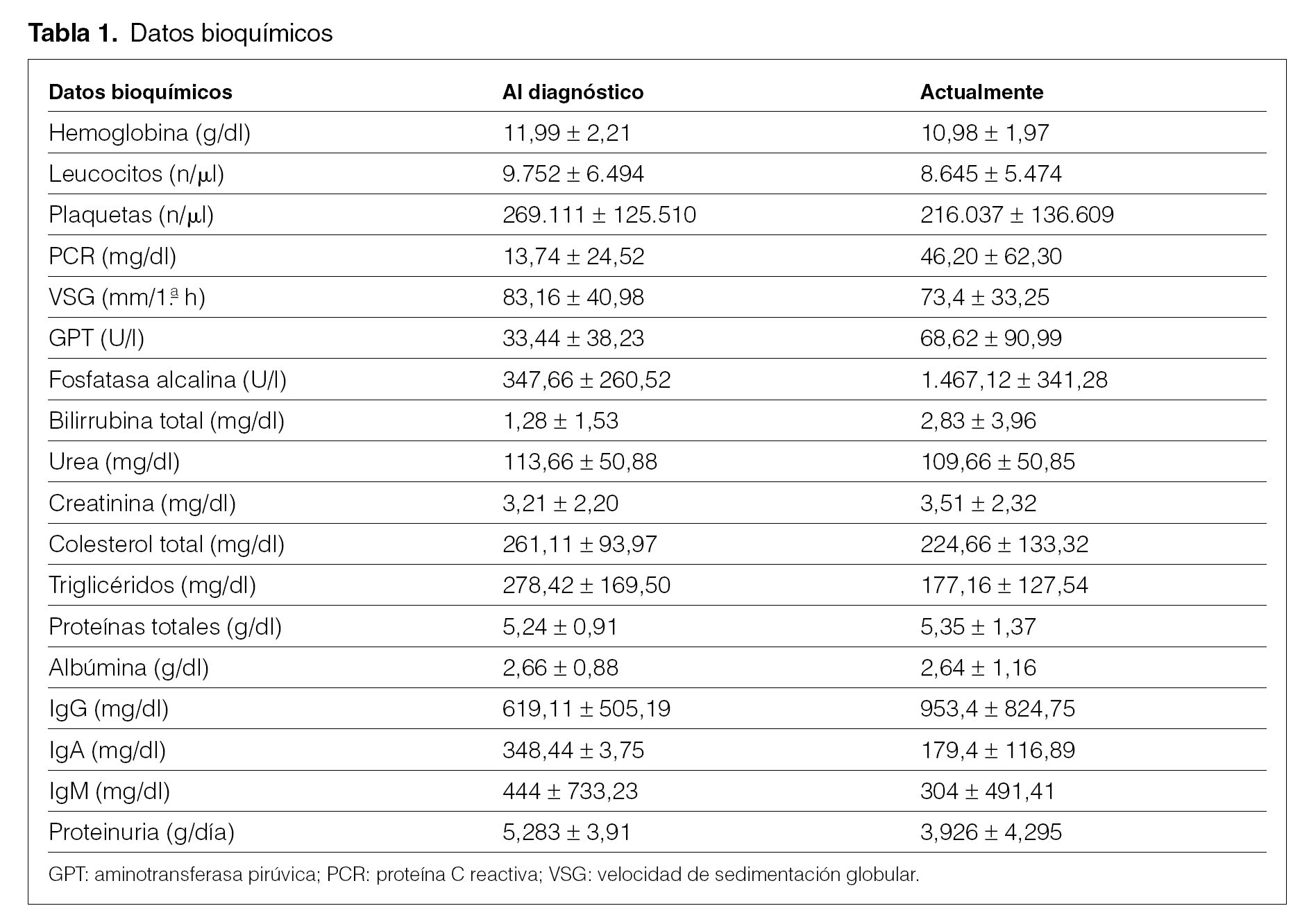
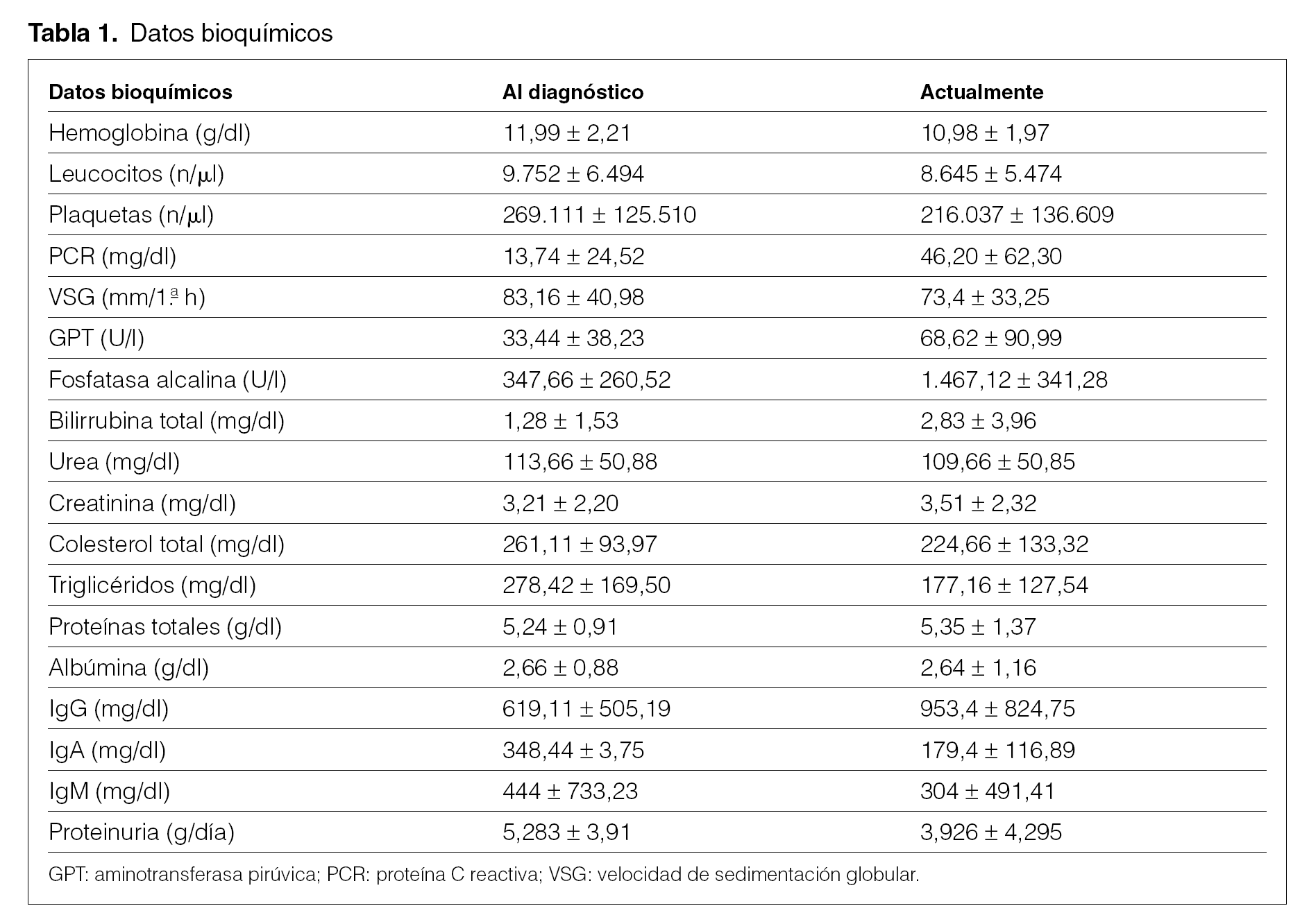
En la tabla 1 se observan los datos más característicos en el momento del diagnóstico y en la actualidad. Destaca la presencia de grave afectación renal, creatinina elevada en 5 pacientes al diagnóstico y en 8 posteriormente, y hepática con patrón analítico de colestasis5. Ocho pacientes presentaron proteinuria en el momento del diagnóstico, 6 de ellos en rango nefrótico. Dos pacientes presentaron picos monoclonales séricos IgM, uno IgA y uno IgG. En la inmunofijación de orina 5 pacientes presentaron proteinuria de Bence-Jones, en 2 predominaron las cadenas k, y en uno, la l.
El riñón fue el órgano más biopsiado para realizar el diagnóstico8. La biopsia gástrica, realizada por la presencia de síntomas digestivos, proporcionó el diagnóstico en 2 pacientes. La punción de médula ósea se realizó de forma habitual8, tras el diagnóstico.
De los 9 pacientes, 4 entraron en programa de hemodiálisis periódica (3 varones y 1 mujer). Cuatro de los pacientes fueron tratados con corticoides junto a inmunosupresores (melfalán, clorambucilo...). Además, a una paciente se le realizó trasplante de médula ósea.
Las principales complicaciones fueron las infecciones5 y las hemorragias digestivas2. Las principales causas de muerte fueron la caquexia y el fracaso multiorgánico3. La media de la supervivencia fue de 13,89 ± 3,03 meses. La supervivencia global fue del 66,6 % a los 2 meses (4 libres de hemodiálisis y 2 en hemodiálisis), y la supervivencia al año fue del 33 % (2 libres de hemodiálisis y 1 en hemodiálisis).
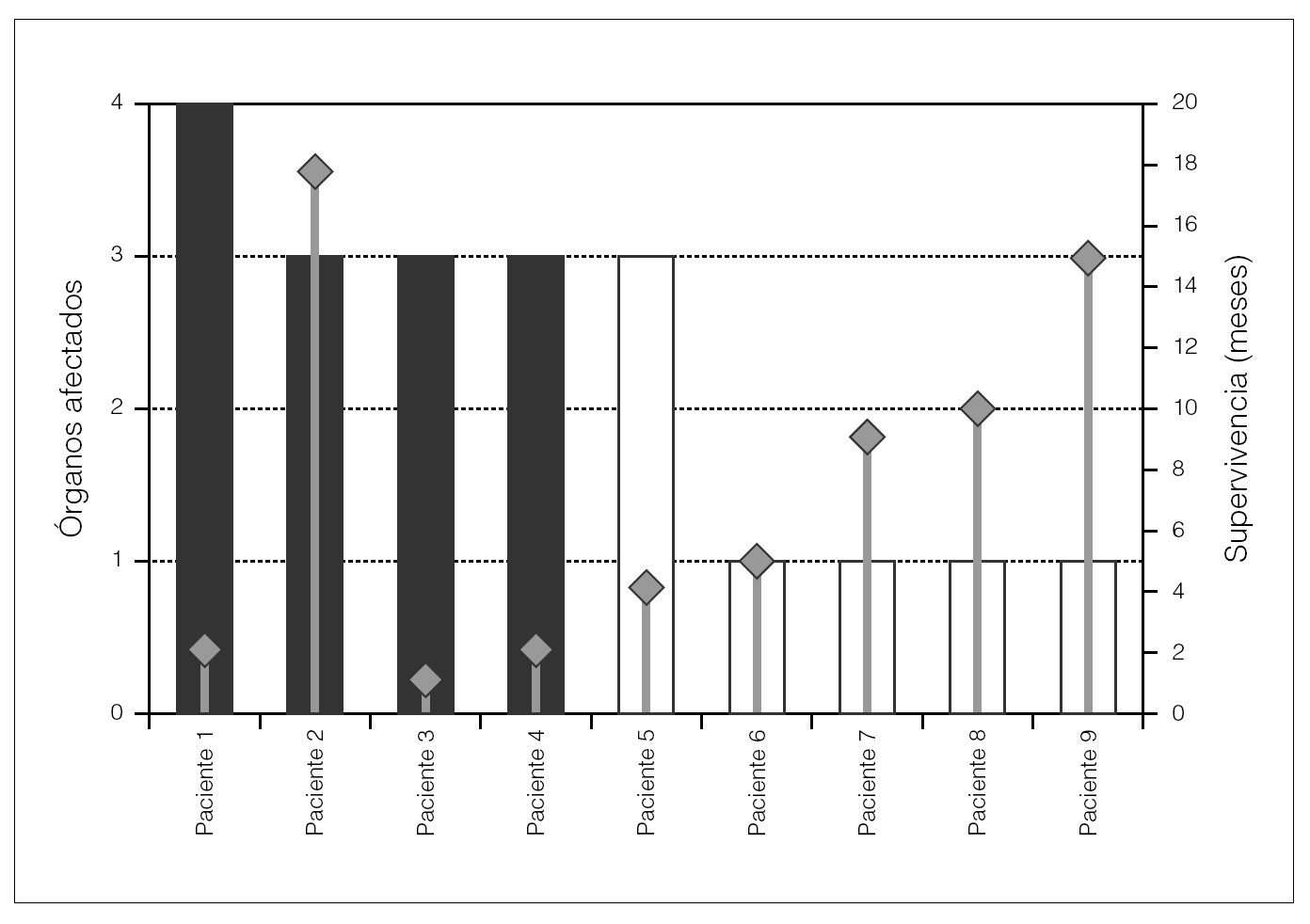
En la figura 1 se aprecia el número de órganos afectados en cada paciente, así como la supervivencia.
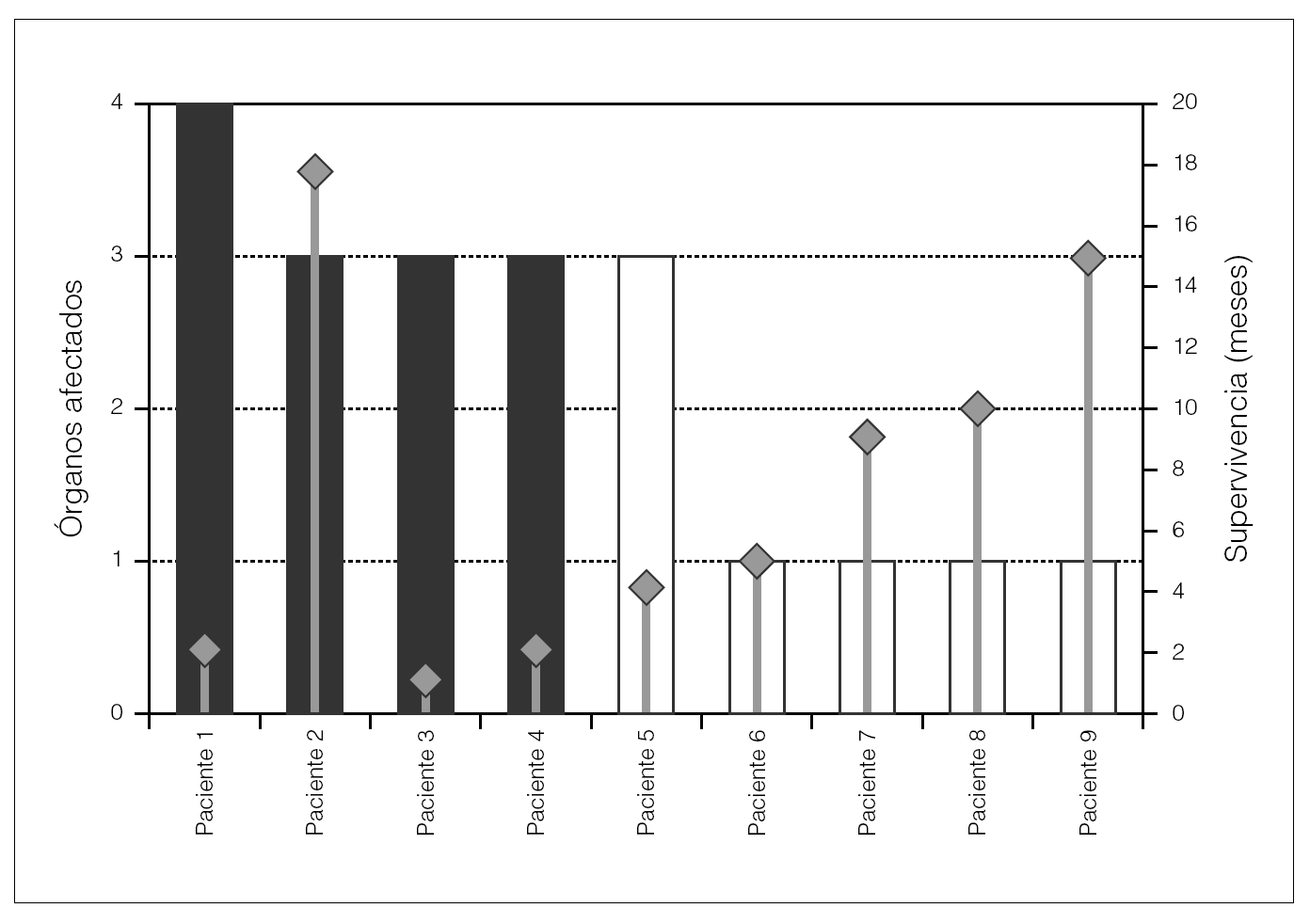
Figura 1. Afectación por órganos (médula ósea, digestivo, corazón y riñón) y supervivencia.
Discusión
La amiloidosis primaria se puede considerar como una discrasia de las células plasmáticas con sobreproducción de componente M, pero con un número pequeño de clones de células plasmáticas en la médula ósea. En nuestro estudio, a diferencia de otros previos, sólo recogemos las amiloidosis de tipo AL que no cumplen criterios de mieloma múltiple. De acuerdo con nuestra casuística, se observa una incidencia de 0,32 casos por cada 100.000 habitantes/ año, mucho menor que la descrita en revisiones estadounidenses, en las que se considera de 0,8 por 100.0003,4. En nuestro hospital predomina la amiloidosis secundaria (1 %)5, a diferencia de otros estudios, donde más de dos tercios eran de tipo AL3,4.
La edad media en el momento del diagnóstico fue de 72,31 años, 8 años superior a la de publicaciones previas3, por lo que puede considerarse una enfermedad que aparece en la edad avanzada. Kyle y Gertz3 afirman que la media de edad ajustada para los varones es más del doble que para las mujeres, mientras que en nuestro estudio no presenta diferencias. Puede existir predilección masculina4, aunque nosotros no la hemos encontrado.
Los síntomas raramente contribuyen a establecer el diagnóstico. Su alto grado de inespecificidad clínica hace que simule un amplio número de enfermedades. Las manifestaciones clínicas afectan a prácticamente todos los órganos, con excepción del sistema nervioso central2. Destacan el cuadro constitucional, los dolores abdominales, sobre todo epigastralgias, y también los vómitos alimentarios de repetición. En la revisión de la clínica Mayo3 sólo el 8 % presentó afectación digestiva. Nosotros observamos dicha afectación en el 66 % de nuestros pacientes, y en 2 de ellos observamos depósitos rojo congo en la biopsia gástrica. Esta prueba se llevó a cabo aprovechando la realización de una gastroscopia para el estudio de epigastralgias de repetición. La presencia de síndrome nefrótico nos indujo a solicitar la tinción referida en las muestras de mucosa gástrica. La amiloidosis digestiva suele demostrarse en casi la totalidad de biopsias gastroduodenales y colorrectales, con la presencia de protrusiones y engrosamiento de los pliegues debidos a la presencia del amiloide.
La afectación renal está presente en el 88,8 % de los casos diagnosticados por nuestro servicio. La proteinuria fue un signo común tanto en la amiloidosis AL como en la AA, llegando en más de la mitad de los casos al rango nefrótico y reduciéndose posteriormente en casi 1 g de media, como consecuencia de la progresión de la insuficiencia renal. Es raro encontrar hipertensión arterial, a pesar de la frecuente alteración de la función renal.
En la biopsia renal se observaron distintas afectaciones, como la glomerular, con depósitos de material rojo congo y PAS positivo en el mesangio y con extensión de éstos hacia las asas capilares; en otros casos se observaron depósitos vasculares, y ocasionalmente glomérulos esclerosos. El 45 % de nuestros pacientes presentó proteinuria de Bence-Jones, porcentaje inferior al descrito por Kyle y Gertz (72 %)3. La inmunohistoquimia anticadenas ligeras k y l fue siempre débilmente positiva. Esto se debe a que en la AL el depósito es sólo de la región variable de cadenas ligeras, y sería necesario el depósito de toda la cadena para una buena captación. Esto último ocurre en la enfermedad de cadenas ligeras2.
Los depósitos en la capa miocárdica se cree que aparecen en más del 50 % de los pacientes, a diferencia de otras formas de amiloidosis, en las que son excepcionales. Nuestros pacientes presentaron engrosamiento del miocardio tanto con aumento de la masa como de la ecogenicidad ecocardiográfica, y en algunas ocasiones se observaron imágenes granulares y centelleantes. Además, se llegaron a observar anomalías en la relajación y patrones de alteración hemodinámica. La disfunción diastólica se considera como el más precoz y la más importante anomalía cardíaca. Los pacientes con insuficiencia cardíaca congestiva presentaron una supervivencia menor a los 6 meses8.
En el electrocardiograma encontramos arritmias y bajos voltajes, en ausencia de patología cardíaca estructural distinta que pudiera explicarlos, y también se describieron patrones de seudoinfarto. Probablemente en estos pacientes existía un trastorno de la conducción a nivel infrahissiano y en el sistema de Purkinje, y éste se debe a la infiltración amiloidea. La prolongación del HV es considerada en un análisis multivariante como factor predictor independiente de muerte súbita7. Simons et al comparan la relación masa-voltaje con otras pruebas no invasivas, como el electrocardiograma o la ecografía, y dan a esta relación una alta sensibilidad para determinar la afectación cardíaca en las amiloidosis7.
La supervivencia a largo plazo es poco frecuente en los pacientes con amiloidosis primaria, y algunas series tienen tasas inferiores a 2 años9,10. Se establece una supervivencia media de 12-18 meses en las amiloidosis no asociadas a discrasias malignas, y todavía es menor en las asociadas a mieloma múltiple11. De 474 pacientes tratados en la clínica Mayo, sólo 30 sobrevivieron más de 10 años10. Los factores de mal pronóstico fueron la insuficiencia cardíaca congestiva, la edad avanzada y valores de creatinina plasmática superiores a 2 mg/dl8. Nuestra media de la supervivencia fue de 14 meses, con una supervivencia global del 66 % a los 6 meses y del 33 % al año.
La afectación cardíaca se ha considerado como primera causa de muerte, por encima de la enfermedad renal8. La mitad de las muertes se consideran de origen cardíaco, a diferencia de nuestro estudio, donde la muerte es resultado de la evolución de esta enfermedad caquectizante con un consiguiente fracaso multiorgánico.
Los fármacos quimioterápicos tratan de disminuir la producción de cadenas ligeras monoclonales. Kyle y Greipp10 no encontraron diferencias en cuanto a supervivencia de los pacientes tratados con prednisona y melfalán frente al grupo control tratado con placebo. Los estudios aleatorizados, como el de Skinner et al, o como otros posteriores de Kyle et al, ya demuestran mejoría de la supervivencia, con medianas de 15 y 8 meses, respectivamente8. Gertz et al observan que el 27 % de sus pacientes responden al tratamiento con agentes alquilantes, con una mediana de supervivencia de los respondedores de 89,4 meses, dato que contrasta con los 14,7 meses de supervivencia de los que no responden. Concluyeron que existe una mejor supervivencia en los pacientes respondedores, y que el número de éstos se relaciona inversamente con los pacientes con aumento de la concentración plasmática de creatinina12. Todavía no existen estudios que evidencien mejores resultados con el tratamiento con prednisona y melfalán a altas dosis y posterior trasplante de médula ósea13, ni con 4'-yodo-4'-desoxidoxorubicina, aunque se esperan resultados prometedores. En nuestra revisión no observamos respuesta en los pacientes tratados con agentes alquilantes y sólo se realizó un trasplante de médula ósea autóloga con buena evolución posterior del paciente. Podemos concluir que la amiloidosis AL es una condición irreversible con afectación multiorgánica y un pobre pronóstico a corto plazo.
Conclusiones
La amiloidosis AL es una enfermedad con elevada morbimortalidad y con constante afectación renal. Fueron frecuentes la afectación digestiva y el patrón analítico de colestasis, la hipotensión postural y las infecciones. Las principales causas de muerte fueron la caquexia y el fallo multiorgánico.
La hemodiálisis constituyó una opción terapéutica adecuada en nuestros pacientes; prolongó la supervivencia en 3 de ellos y permitió que uno recuperara la función renal.