






Los trastornos relacionados con mutaciones del gen IRF6, comprenden desde una afectación casi asintomática con la única presencia de hoyuelos labiales que son la manifestación más sutil del síndrome de van der Woude, hasta manifestaciones congénitas graves que incluyen anomalías faciales, musculoesqueléticas y genitourinarias que corresponden al síndrome de pterigium poplíteo.
Pese a que existe cierta relación fenotipo-genotipo entre las mutaciones del gen IRF6, estas tienen una penetrancia incompleta y una expresión variable, inter e intrafamiliar.
The disorders related to IRF6 encompass a spectrum from an almost asymptomatic affectation, with the only presence of isolated lip pits, which are a mild presentation of van der Woude syndrome, to the presence in the other extreme, of congenital manifestations that include facial anomalies, musculoskeletal and genitourinary malformations, corresponding to popliteal pterygium syndrome.
Although there is a certain phenotype-genotype relationship between mutations of the IRF6 gene, such mutations have incomplete penetrance and variable inter- and intra-familial expression.
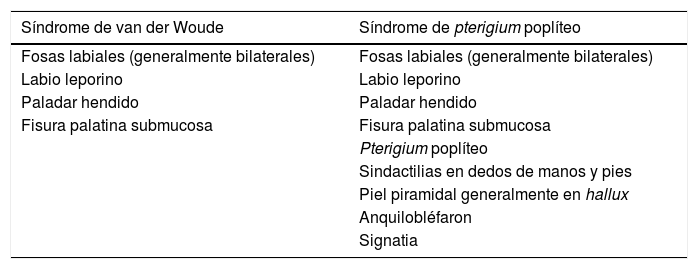
El síndrome de van der Woude (SVW) (OMIM #119300) con una frecuencia entre uno y 9 por cada 100.000, se puede presentar en su forma más leve como fositas labiales y en su forma más grave como paladar hendido con o sin labio leporino, mientras que el síndrome de pterigium poplíteo (SPP) (OMIM # 119500) con una incidencia de uno cada 300.000, presenta manifestaciones congénitas mucho más graves a nivel facial, musculoesqueléticas y genitourinarias (tabla 1).
Variabilidad fenotípica del gen IRF6
| Síndrome de van der Woude | Síndrome de pterigium poplíteo |
|---|---|
| Fosas labiales (generalmente bilaterales) | Fosas labiales (generalmente bilaterales) |
| Labio leporino | Labio leporino |
| Paladar hendido | Paladar hendido |
| Fisura palatina submucosa | Fisura palatina submucosa |
| Pterigium poplíteo | |
| Sindactilias en dedos de manos y pies | |
| Piel piramidal generalmente en hallux | |
| Anquilobléfaron | |
| Signatia |
Listado de las diferentes manifestaciones en alteraciones del gen IRF6 agrupadas bajo el síndrome de van der Woude o del síndrome de pterigium poplíteo.
El SVW presenta en la mayoría de los casos fosas labiales aisladas, aunque puede presentar alteraciones del paladar, úvula bífida, anquiloglosia, anomalías en las extremidades (pliegues de la piel y/o sindáctila) y pérdida de audición neurosensorial.
El SPP se caracteriza por la presencia de unas membranas de piel que se extienden desde la tuberosidad isquiática a los talones con contracturas que perjudican la movilidad, denominadas pterigium poplíteo y piel recubriendo en disposición piramidal las uñas generalmente del hallux, esta última característica es patognomónica del síndrome. Otras manifestaciones asociadas incluyen fosas labiales, paladar hendido con o sin labio leporino, anquilobléfaron (sinéquias filiformes conectando los párpados superiores e inferiores) o signatia (cuando las sinéquias se presentan conectando el maxilar superior y el inferior), criptorquidia y escroto bífido en varones e hipoplasia de labios mayores en mujeres.
Ambos síndromes no alteran la capacidad cognitiva.
El SPP es provocado por mutaciones del gen IRF6. El SVW en el 70% de los casos se debe a mutaciones del gen IRF6, en un pequeño porcentaje a microdelecciones del mismo IRF6 y el resto a mutaciones en otros genes, como GRHL3 o de etiología desconocida4. Las mutaciones del gen IRF6 tienen penetrancia incompleta y expresión variable para ambos síndromes.
El gen IRF6 está situado en la región cromosómica 1q32, y está compuesto por 9 exones y 8 intrones. Codifica por la proteína IRF6 que pertenece a la familia de los interferones (reguladores de los factores transcripción) y, aunque su función es actualmente poco conocida, se ha asociado al desarrollo de la epidermis y a la regulación del desarrollo craneofacial (fig. 1).
Caso clínico
Presentamos el caso de una gestante, la ecografía fetal realizada en el primer trimestre detecta inicialmente una baja movilidad de las extremidades inferiores, y en ecografías subsiguientes se detecta la presencia de labio leporino bilateral. Se realizan los siguientes estudios complementarios:
- -
Estudio de aneuploidías 13, 18 y 21, mediante la técnica quantitative fluorescence polymerase chain reaction (qf-PCR) kit Devyser Compact v3, con resultado compatible con la normalidad.
- -
Estudio de microdeleciones y microduplicaciones mediante array de hibridación genómica comparada (Array-CGH) qChip Pre v1.1, 60.000 oligonucleótidos Q-genomics, con resultado compatible con la normalidad.
- -
Estudio de malformaciones fetales mediante resonancia magnética nuclear (RMN), en el que se visualiza labio leporino bilateral con afectación del paladar óseo. El resto de estructuras están dentro de la normalidad.
En los estudios posteriores no se detectan otras alteraciones morfológicas fetales, ni la causa de la baja movilidad de las piernas. Frente al cuadro polimalformativo y por el potencial riesgo que conlleva de trastornos del neurodesarrollo, la pareja decide finalizar el embarazo.
El estudio anatomopatológico corresponde a un feto de sexo masculino de 21 semanas con labio leporino bilateral (completo en la izquierda y parcial en la derecha) y paladar hendido (fig. 2A), hipertrofia gingival (fig. 2B), ausencia de saco escrotal y pterigium poplíteo bilateral con flexión de las extremidades inferiores (fig. 2C), pie varo derecho, hipoplasia de las uñas en el primer y segundo dedo del pie y en el segundo dedo de la mano derecha con redundancia de la piel ungueal en forma de pirámide (fig. 2D).

A partir de la autopsia fetal se determina la sospecha diagnóstica de SPP por la presencia de piel ungueal en forma de pirámide y la presentación de contracturas en las extremidades inferiores, promoviendo la realización del análisis de secuenciación de la región codificante de los exones 3 al 9 del gen IRF6 utilizando la nomenclatura del Human Genome Variation Society mediante kit específico AmpFISTR® Profiler Plus® kit d’Applied Biosystems.
El estudio de secuenciación detecta una mutación en heterocigosis c.250C>T (p.Arg84Cys) en el exón 4. Esta mutación por cambio de sentido (missense) da lugar a un cambio de una arginina por una cisteína en la posición 84, provocando un cambio de un único aminoácido y generando una proteína anómala. La mutación p.Arg84C ya ha sido reportada como patogénica previamente en la literatura científica1–3. La ausencia de la mutación en los progenitores determina que la variante es de novo.
En todo el proceso de estudio se han seguido los protocolos de actuación del hospital y se ha preservado la privacidad de la pareja.
DiscusiónActualmente conocemos que diferentes mutaciones del gen IRF6 pueden provocar de forma autosómica dominante que los individuos permanezcan casi asintomáticos o que presenten manifestaciones clínicas de SVW o SPP. Observándose cierta correlación entre la tipología y la localización de la mutación a lo largo del gen, y el tipo de manifestaciones que presentará el individuo. En el SPP generalmente es causado por mutaciones missense localizadas en el dominio de unión al ADN de los exones 3 y 4, mientras que las mutaciones causantes del SVW son generalmente mutaciones de missense localizadas en los exones 3 y 4 no asociadas sitios de unión del ADN y en los exones 7 y 9. Sin embargo, esta asociación genotipo-fenotipo no es absoluta, ya que actualmente se han identificado unos pocos pacientes con mutaciones de cambio de sentido que estando localizadas en el dominio de unión del ADN y en el exón 4, y presentan manifestaciones del SVW1.
En nuestro caso, la mutación p.Arg84Cys es una mutación localizada en el exón 4 en el dominio de unión a ADN en una región altamente conservada del gen que provoca que su penetrancia sea casi completa aunque su expresión pueda ser variable, como se demuestra en el estudio de Renata et al. del año 2009. En este estudio de las familias con mutación p.Arg84Cys, 3 individuos presentaron SVW y 18 individuos SPP, incluso encontrando casos de SVW y SPP dentro de la misma familia1.
ConclusiónNo es posible determinar una correlación entre las diferentes mutaciones de IRF6 y la presencia y gravedad de las manifestaciones, pero los datos más consistentes atribuyen que las mutaciones que provocan una pérdida total o parcial de la función del gen se manifiestan como SVW, y las mutaciones de efecto dominante negativo se les atribuye el SPP. El espectro fenotípico del SVW hasta el SPP, incluyendo la superposición que existe en algunas de estas manifestaciones, sugiere las posibles contribuciones de eventos estocásticos y modificadores genéticos del gen IRF64. Así, no podemos predecir con exactitud las manifestaciones que provocará una mutación del gen IRF6 en diferentes miembros de una misma familia.
Podemos concluir que ambas entidades son extremos opuestos del espectro de trastornos provocados por mutaciones del gen IRF6. Así el asesoramiento genético a parejas con embarazos a riesgo o presencia de mutación del gen IRF6 debe considerar el riesgo de presentación para ambos síndromes independientemente de la localización, la tipología de mutación o la presentación previa en la familia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Comunicación escrita en el European Human Genetics Conference 2016, Barcelona. Título del póster presentado: «The process of genetic counselling and the genotype-phenotype correlation in a popliteal pterygium syndrome: a case report».
