






La sitosterolemia es una enfermedad autosómica recesiva y muy infrecuente. Su principal característica es que se presenta una mayor absorción y una disminución en la excreción de esteroles, lo que conlleva a que se depositen en los tejidos. Está dada por mutaciones en el gen ABCG5 o en el ABCG8, que se encuentran en el cromosoma 2p21. En esta nota clínica se describen los dos primeros pacientes con sitosterolemia familiar descritos en Colombia, hermanos, uno de ellos con nódulos xantomatosos en las extremidades como único síntoma, y el otro, completamente asintomático. Se realizaron estudios genéticos como prueba diagnóstica en ambos pacientes, donde se pudo identificar una variante homocigota patogénica en el gen ABCG8 en el primer caso (sintomático) y una variante heterocigótica en el gen ABCG8 en el segundo caso (asintomático); el primer paciente ha respondido al tratamiento con ezetimiba. En conclusión, los xantomas deben ser estudiados a profundidad en la edad pediátrica, ya que pueden ser el único signo ostensible de enfermedades tan complejas y hereditarias como la sitosterolemia familiar, la cual puede ser controlada y así evitar las complicaciones cardiovasculares propias de la enfermedad.
Sitosterolemia is an autosomal recessive and very rare disease. Its main characteristic is that there is a greater absorption and a decrease in the excretion of sterols, which leads to them being deposited in tissues. It is given by mutations in the ABCG5 or ABCG8 genes found on chromosome 2p21. In this clinical note, we describe the first two patients with familial sitosterolemia described in Colombia, brothers, one of them with xanthomas in extremities as the only symptom, and the other, completely asymptomatic. Genetic studies were performed as a diagnostic test in both patients, where a pathogenic homozygous variant could be identified in the ABCG8 gene in the first case (symptomatic), and a heterozygous variant in the ABCG8 gene in the second case (asymptomatic); the first patient has responded to treatment with ezetimibe. In conclusion, xanthomas should be studied in depth in pediatric age as they may be the only visible sign of such complex and hereditary diseases as familial sitosterolemia, which can be controlled and prevent cardiovascular complications of the disease.
La sitosterolemia se considera un raro trastorno hereditario autosómico recesivo del almacenamiento de los lípidos, y su origen se da por mutaciones homo o heterocigóticas en los genes ABC que codifican los transportadores de salida del heterodímero hepático e intestinal G5 y G8 localizados en el cromosoma 2p211. Los genes ABCG5 y ABCG8 codifican para los transportadores expresados en el tracto biliar y el enterocito, cuya función es transportar esteroles fuera de las células a la luz intestinal o hacia la bilis. Se caracteriza por una mayor absorción y una disminución en la excreción biliar de esteroles vegetales y colesterol, dando como resultado concentraciones elevadas de esteroles de origen vegetal, como el sitosterol, el campesterol y el estigmasterol, teniendo como consecuencia su depósito en los tejidos2. Las personas sanas absorben solo aproximadamente el 5% del promedio de 200 a 300mg de esteroles vegetales consumidos cada día. Casi todo el sitosterol absorbido se excreta rápidamente en la bilis, de modo que solo quedan trazas de sitosterol y otros esteroles vegetales en la sangre. Por el contrario, las personas con sitosterolemia absorben entre el 15 y el 60% del sitosterol ingerido y excretan solo una fracción en la bilis1.
Dentro de sus manifestaciones clínicas se puede identificar la presencia de xantomas cutáneos, aterosclerosis, artritis, hepatomegalia y esplenomegalia, glóbulos rojos de tipo lipídico, macroplaquetas y trombocitopenia3. Además de las medidas dietarias, donde es fundamental evitar los alimentos que contienen fitoesteroles, el uso del inhibidor de la absorción de esteroles, ezetimiba, posiblemente en combinación con colestiramina, una resina fijadora de ácidos biliares, es la opción más eficaz para su tratamiento3. El diagnóstico de la sitosterolemia en la infancia es un verdadero desafío, dado que, sin un análisis preciso de los esteroles plasmáticos, es más fácil diagnosticarla erróneamente como hipercolesterolemia familiar4.
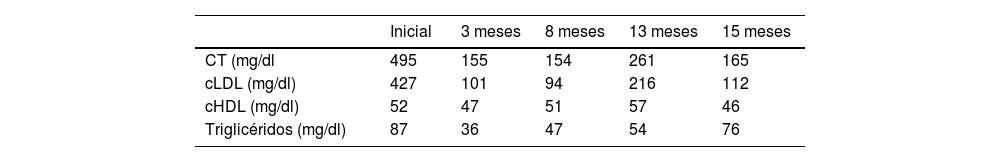
Caso clínico 1Paciente de 7 años de edad que consulta por nódulos xantomatosos agrupados, localizados en las rodillas, de tres años de evolución, no dolorosos, sin otro síntoma asociado inicialmente (fig. 1). Habían realizado pruebas de laboratorio que revelaron los siguientes valores lipídicos; colesterol total (CT): 495mg/dl, y colesterol de lipoproteínas de baja densidad (cLDL): 427mg/dl. Inicialmente se interpretó como un caso de hipercolesterolemia familiar (según los criterios de la red de clínicas de lípidos holandesa Dutch Lipid Clinic Network [DLCN] para el diagnóstico de hipercolesterolemia familiar; su puntaje fue de 14, a sabiendas que un resultado >8 se considera como una certeza en el diagnóstico), por lo que recibió tratamiento con atorvastatina durante 6meses, con disminución en los valores de lípidos séricos. Sin embargo, aparecieron nuevos nódulos xantomatosos localizados en los codos y en retropiés a nivel los tendones de Aquiles (fig. 1). No presentaba antecedentes familiares de hipercolesterolemia y/o de accidentes cardio o cerebrovasculares. Posterior a una exhaustiva búsqueda, tiempo después de la sospecha inicial de hipercolesterolemia familiar, indagando nuevamente sobre los antecedentes familiares, se descubrió que el abuelo paterno falleció a los 42años por un evento coronario agudo.
Xantomas en codos. B)Xantomas en nudillos. C)Xantomas en retropié, a nivel de tendón de aquiles bilateral. D)Xantomas en ambas rodillas.")
El examen físico mostró un peso de 24,5kg, talla de 124cm, puntajeZ (P/E: 0,28, T/E: 0,34, IMC: 0,08kg/m2), parámetros antropométricos adecuados para la edad. La presión arterial fue normal en ambas extremidades. Se identificaron múltiples nódulos xantomatosos en ambas rodillas, en codos, en el segundo metacarpiano de ambas manos y en retropié, que comprometían ambos tendones de Aquiles (fig. 1). No se identificaron adenomegalias.
Los resultados de laboratorio iniciales revelaron CT: 495mg/dl, triglicéridos: 87mg/dl, colesterol de lipoproteínas alta densidad (cHDL): 51mg/dl, y cLDL: 427mg/dl (tabla 1). No se encontraron anomalías en la función hepática, en la renal, ni en la glucosa en sangre.
Se realizó ecocardiograma doppler color, en el que no se identificaron patologías cardíacas. La ecografía abdominal fue normal. El extendido de sangre periférica reportó: serie roja con hipocromía, anisocitosis ligera con microcitos y poiquilocitosis ligera con dianocitos, serie blanca y roja normales en número y en morfología. Se realizó la biopsia de una de las lesiones localizada en la rodilla, reportando población de histiocitos con CD68, concluyendo una histiocitosis de células no Langerhans.
Las pruebas genéticas se realizaron mediante secuenciación y análisis de variantes en número de copia (CNV) mediante NGS en genes relacionados con dislipidemias primarias en el centro de genética avanzada Gencell. Se evaluaron 26 genes: ABCG5, ABCG8, APOA1, APOA2, APOA5, APOB, APOC2, APOC3, APOE, APTX, CREB3L2, CYP27A1, EPHX2, GHR, GPD1, GPIHBP1, ITIH4, LDLR, LDLRAP1, LIPA, LIPI, LMF1, LPL, LRP6, PCSK9 y PPP1R17. Se identificó una variante homocigota patogénica en el gen ABCG8 que consiste en la duplicación de 11 nucleótidos (GGGTGAGCGCA) entre las posiciones 647 y 657 del exón 5/13 del ADNc del gen (c.647_657dup), que a nivel de la proteína produce el cambio frameshift de corrimiento del marco de lectura, que lleva a un codón de parada prematuro en el aminoácido 256 (p.Arg220 ValfsTer37) en una proteína de 673 aminoácidos.
Después del diagnóstico definitivo, al paciente1 se le administró: ezetimiba 10mg/día y rosuvastatina 10mg/día, acompañado de una dieta hipograsa baja en esteroles vegetales y actividad física regular. Se realizó seguimiento regular en clínicas pediátricas, cada 6-12semanas. Luego de 15meses de este tratamiento, teniendo en cuenta la disminución de las lesiones y los valores séricos normales de lípidos, se decidió suspender la rosuvastatina, y hasta la fecha continúa recibiendo ezetimiba.
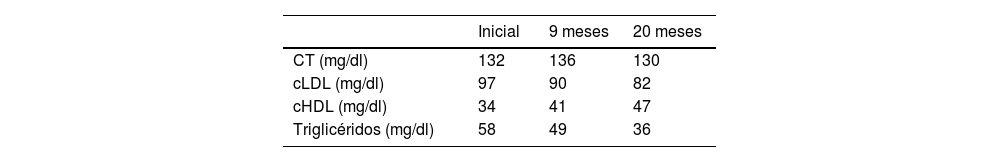
Caso clínico 2La hermana del paciente1 presenta una edad de 5años, posterior al diagnóstico definitivo del hermano; se le tomaron controles de perfil lipídico anualmente durante tres años, donde no se han evidenciado alteraciones en los resultados. Sin embargo, debido a la consanguineidad, se realizó el análisis genético mediante secuenciación y análisis de CNV mediante NGS en genes relacionados con dislipidemias primarias en el centro de genética avanzada Gencell. Se evaluaron 26 genes: ABCG5, ABCG8, APOA1, APOA2, APOA5, APOB, APOC2, APOC3, APOE, APTX, CREB3L2, CYP27A1, EPHX2, GHR, GPD1, GPIHBP1, ITIH4, LDLR, LDLRAP1, LIPA, LIPI, LMF1, LPL, LRP6, PCSK9 y PPP1R17. Se identificó la variante heterocigótica c.647_657dup en el gen ABCG8. Los parámetros de lípidos de los familiares de primer grado se representan en la figura 2 y en la tabla 2. Las medidas antropométricas corresponden a un peso de 16,6kg, talla de 110cm, puntajeZ (P/E: −0,89, T/E: −0,26, IMC: −1,13kg/m2), con riesgo de delgadez, pero el resto de parámetros antropométricos son adecuados para la edad. Su examen físico no mostró hallazgos patológicos, ni tampoco se identificó la presencia de xantomas. A día de hoy no ha desarrollado manifestaciones clínicas asociadas. Teniendo en cuenta la ausencia de síntomas y el diagnóstico reciente, aún no se le han realizado estudios adicionales al perfil lipídico ni se le ha administrado tratamiento.
Discusión
La sitosterolemia es un trastorno mendeliano raro (autosómico recesivo) del metabolismo de los lípidos, caracterizado por una mayor absorción y una disminución de la excreción biliar, principalmente de esteroles vegetales (sitosterol, campesterol y estigmasterol) y colesterol, lo que resulta en un aumento de sus concentraciones séricas4. En condiciones normales, solo el 5% de los fitoesteroles que se absorben permanecen en el organismo. Fue descrito por primera vez en 1974 por Bhattacharyya y Connor5. Este trastorno se relaciona con la aparición de nódulos xantomatosos, que por lo general se encuentran en talones, rodillas, codos y glúteos. Está asociado a complicaciones como aterosclerosis prematura, enfermedades cardiovasculares, alteraciones hematológicas como anemia hemolítica, estomatocitosis, plaquetas gigantes, esplenomegalia, artralgias y artritis6. Todas estas características se asemejan a la hipercolesterolemia familiar (HF).
Los pacientes heterocigotos son normales clínicamente y pueden presentar niveles levemente elevados de fitoesteroles en plasma. Los niveles de colesterol pueden estar normales; sin embargo, lo usual es que estén significativamente elevados, incluso en pacientes con lactancia materna, aparentemente antes de que se eleven en sangre los fitoesteroles7.
La acumulación de esteroles vegetales en estos pacientes en particular es la principal causa de aterogénesis. Se han descrito casos de infarto agudo de miocardio y muerte súbita a edades muy tempranas a causa de la aterosclerosis secundaria, al igual que paraplejía por compresión del canal medular debido a xantomas e insuficiencia suprarrenal8.
Hay mutación de los genes ABCG5 o ABCG8 del cromosoma 2p21 que codifican para dos transportadores intestinales, denominados ABCG5 y ABCG8. Estos genes codifican dos proteínas (Sterolin-1 y Sterolin-2), las cuales forman un transportador de heterodímeros ubicado en el retículo endoplásmico de la membrana del borde en cepillo de los enterocitos y la membrana canalicular de los hepatocitos, y aquí es donde actúan interviniendo en el transporte del esterol absorbido desde el espacio intracelular al extracelular9.
Las manifestaciones cutáneas de la HF son la clave clínica para llegar al diagnóstico precoz. Estas incluyen los xantomas cutáneos, los xantomas tuberosos y los xantomas tendinosos, estos dos últimos con una fuerte asociación con la HHF cuando se inician en la infancia. Estas manifestaciones cutáneas pueden revertirse de manera lenta al corregir los niveles de colesterol en sangre, pero muchas veces requieren extirpación quirúrgica. El diagnóstico se confirma con el estudio genético; si este no estuviera disponible o si el caso plantea dudas, el diagnóstico puede apoyarse en el cuadro clínico, la historia familiar y el laboratorio. Actualmente, el diagnóstico de sitosterolemia se basa principalmente en el nivel de esteroles vegetales séricos y la secuenciación de los genes. Es cierto que estos esteroles vegetales no logran distinguirse del colesterol por los métodos normalmente utilizados, y en consecuencia es necesario realizar cromatografía gaseosa y espectrometría de masas (CG/EM) para identificarlos. En las instituciones prestadoras de servicios en salud es poco usual que se cuente con este tipo de tecnología, por lo que muy a menudo se diagnostica erróneamente la sitosterolemia como hipercolesterolemia. La principal técnica de diagnóstico, y más accesible que la CG/EM, es la secuenciación de genes. Se ha encontrado que las mutaciones en ABCG5 duplican en frecuencia las de ABCG8 en los casos de sitosterolemia. Los principales sitios donde se observa la mutación del gen ABCG5 son mutaciones de exón; sin embargo, hay un número reducido de pacientes que presentan las mutaciones de intrón10.
Con base en las últimas guías publicadas, el tratamiento de la HHF debe ser instaurado de manera precoz y su objetivo es reducir los niveles de cLDL, idealmente a valores <70mg/dl, para evitar la ateromatosis y el daño cardiaco irreversible. A todos los pacientes se les deben indicar modificaciones en el estilo de vida, con dieta rica en frutas, vegetales y granos enteros, baja en grasas saturadas y colesterol, asociada a la actividad física. También se hace mención a una dieta pobre en esteroles e hipograsa. La restricción dietética de esteroles vegetales debería ser la estrategia de primera línea, pero una dieta como la mediterránea es adecuada y no es pobre en grasa (supera el 35% de valor calórico total (VCT) a expensas de la grasa monoinsaturado (MI), cuya fuente es el aceite de oliva).
El manejo farmacológico clásico se basa en el uso de ezetimiba y colestiramina. La combinación de estatina y ezetimibe no suele ser suficiente, y es ineficaz si no hay actividad residual del receptor de LDL. Los pacientes con sitosterolemia generalmente no responden a las estatinas porque la actividad de la HMG-CoA reductasa ya se inhibe al máximo en la sitosterolemia. Sin embargo, las estatinas son eficaces para reducir el cLDL, al menos en algunos pacientes sitosterolémicos2. Sin embargo, no reduciría los niveles séricos de esteroles vegetales y, por lo tanto, la terapia con estatinas por sí sola no es un tratamiento apropiado para la sitosterolemia. En definitiva, es posible la introducción de estatina asociada a ezetimiba, pero no parece adecuada, en ausencia de secundarismos, su retirada ante una mejora del perfil lipídico. Por último, es importante el consejo genético para diagnosticar la variante heterocigota en otros familiares y considerar el riesgo en la descendencia. En los casos presentados se hace el primer reporte de sitosterolemia en pacientes pediátricos en Colombia, y aunque no fue posible realizar la determinación de esteroles séricos, el cuadro clínico y la prueba genética llevaron al diagnóstico y se inició tratamiento con el esquema clásico recomendado asociado a modificaciones en el estilo de vida, con buen control clínico y de laboratorio.
Estos casos resaltan la importancia del conocimiento y la pesquisa oportuna de los xantomas tuberosos como primera manifestación de la HHF. Los primeros en detectar sus manifestaciones clínicas serán los médicos de atención primaria, los pediatras y los dermatólogos, por lo que resulta esencial el reconocimiento, la derivación oportuna y el manejo multidisciplinario para prevenir la aterosclerosis acelerada y la muerte precoz en el paciente y en sus familiares.
FinanciaciónNinguna.
Conflicto de interesesLos autores declaran no tener conflictos de intereses aplicables a esta publicación.
Al caso índice y sus familiares estudiados, que dieron su consentimiento para la publicación anónima.