





La enfermedad de von Willebrand es el trastorno hereditario más frecuente de las proteínas de la coagulación en los seres humanos. Existen 3 tipos: 1, 2A, 2B, 2N, 2M, y 3. Está asociada a mutaciones en el cromosoma 12, en la región p13.2, que codifica para el factor de von Willebrand (VWF), el cual se sintetiza en las células endoteliales y megacariocitos.
DiscusiónLa biología molecular ha permitido la caracterización del gen del VWF, adquiriendo un papel importante en el diagnóstico la enfermedad de von Willebrand así como en la investigación de alteraciones en otros genes, que pueden estar involucrados en la regulación de la síntesis, procesamiento y secreción del VWF. Sin embargo, aún no se han integrado las estrategias de biología molecular entre las pruebas de diagnóstico disponibles.
El análisis de los multímeros del VWF es una metodología que cumple con las características para el diagnóstico, pero no es fácil de estandarizar. Tomando en consideración que aún en los centros de tercer nivel en nuestro país los enfermos de von Willebrand no cuentan con un diagnóstico definitivo, es necesario implementar estas metodologías para su estudio y mejorar su diagnóstico.
ConclusionesLa enfermedad de von Willebrand es heterogénea debido a los mecanismos moleculares que producen los distintos fenotipos clínicos y de laboratorio. En México existen pocos trabajos relacionados con esta enfermedad, por ello es fundamental realizar un estudio integral que incluya aspectos clínicos, pruebas de laboratorio básicas y especiales, para establecer el diagnóstico correcto, desarrollar nuevos enfoques terapéuticos, y así ofrecer atención médica y asesoramiento genético adecuados.
Von Willebrand disease is the most common inherited disorder of the coagulation proteins in humans. There are three types: 1, 2A, 2B, 2N, 2M and 3. It is associated with mutations on chromosome 12 in the region p13.2, encoding the von Willebrand factor (VWF), which is synthesized in endothelial cells and megakaryocytes.
DiscussionThe VWF gene has been characterised using molecular biology techniques, which have acquired an important role in diagnosis von Willebrand disease, as well as in the investigation of alterations in other genes, which may be involved in regulating the synthesis, processing, and secretion of VWF. However, there are still no strategies to integrate the molecular biology diagnostic tests available.
Analysis of VWF multimers is a methodology that meets the characteristics for diagnosis, but it is not easy to standardise. Considering that even in tertiary centres in our country, von Willebrand patients do not have a definitive diagnosis, it is necessary to implement these methodologies to study and improve diagnosis.
ConclusionsVon Willebrand disease is highly heterogeneous due to the molecular mechanisms that produce the various clinical and laboratory phenotypes. In Mexico there are few studies related to this disease; therefore it is essential to conduct a comprehensive study including clinical, basic, and special testing laboratory tests, in order to establish a correct diagnosis, develop new therapeutic approaches, and offer the appropriate medical care and genetic counselling.
Los trastornos de la coagulación son anormalidades hemostáticas hereditarias y algunos de ellos pueden presentar considerables dificultades diagnósticas y de tratamiento. La enfermedad de von Willebrand es un trastorno hereditario que se caracteriza por hemorragias mucocutáneas de intensidad variable, que afecta primordialmente la hemostasia primaria en la interacción plaquetaria, factor de von Willebrand (von Willebrand factor, VWF) y endotelio. Implica cambios en la estructura, función o concentración del VWF, una proteína plasmática secretada por las células endoteliales que circula en el plasma1.
De acuerdo con Vischer y de Moerloose, Erik Adolf von Willebrand en 1926 describió un grave trastorno hemorrágico denominado seudohemofilia en una familia que presentaba tiempos de sangrado prolongados a pesar de tener un conteo plaquetario normal1,2. Posteriormente se demostró que este trastorno se relaciona con disminución de la actividad procoagulante del factor VIII de la coagulación (FVIII), que puede compensarse mediante la infusión de plasma o fracciones de plasma, lo que hace evidente que la enfermedad se debe a la falta de un factor plasmático3. En 1971 fue descrito que el FVIII y el VWF eran proteínas distintas, y en 1975 Gralnick y Coller4 caracterizaron el VWF. Este descubrimiento se acompañó de una nueva prueba de laboratorio que emplea ristocetina para evaluar la función plaquetaria en esta enfermedad4,5 y, en 1985, la naturaleza diferente del VWF quedó definitivamente demostrada al describir la secuencia del gen del VWF5,6.
Factor de von WillebrandEl VWF es una proteína multimérica que se sintetiza en células del endotelio vascular, megacariocitos y plaquetas con vida media de 12h, es codificado en un gen de 52 exones (178Kb) localizado en 12p13.2, y transcribe un mRNA de 8.8Kb (2813 aminoácidos [aa]). Actualmente se conocen más de 160 variantes normales en la estructura del gen. Su transcripción está regulada por factores de transcripción específicos del tipo de célula (proteínas GATA y ETS), y también existen varios elementos transcripcionales represivos en la secuencia ascendente del gen (fig. 1)7–9. Por otro lado, en el cromosoma 22 existe una copia parcial (seudogén) de los exones 23 al 34 de la secuencia del cromosoma 12; este remanente evolutivo no funcional muestra una divergencia de secuencia del 3% respecto del gen del cromosoma 12 y parece haber sido generado más o menos durante la época en la que los primates de orden mayor se diferenciaron de los monos, lo cual parece complicar su análisis10. El VWF es una glucoproteína (Gp) plasmática; se calcula que el 75-85% del VWF que circula libre en el plasma deriva del endotelio, en tanto que el 15-25% restante se encuentra almacenado en las plaquetas circulantes que se originan a partir del megacarocito1,11.

Representación del gen, el transcrito y la proteína del factor de von Willebrand. Se observa la localización del gen del factor de von Willebrand en el cromosoma 12, el mRNA maduro y el procesamiento de la proteína, desde el pre-pro-factor de von Willebrand, hasta la formación de los multímeros de alto peso molecular.
Durante la síntesis del VWF se forma la proteína llamada pre-pro-VWF; es un producto inicial de 300-350KDa (∼2813 aa), que contiene un péptido señal de 22aa, un propéptido de 741aa y una proteína madura de 2050aa. La estructura de la proteína del VWF está formada por varios dominios repetidos en el orden D1-D2-D’-D3-A1-A2-A3-D4-B1-B2-B3-C1-C2-CK. Los dominios D1, D2, D’ y D3 participan en la regulación del proceso de formación de multímeros, y las regiones D’ y D3 también median la unión con el FVIII. Tanto el dominio A1 como el A3 poseen propiedades de unión al colágeno. Los sitios donde el VWF se une a las plaquetas son: en el dominio A1 al receptor plaquetario Gp Ib/IX (GpIb/IX), y en el dominio C2 al receptor Gp IIb/IIIa (GpIIb/IIIa). De tal forma que cada monómero de VWF posee dominios que permiten a la proteína unirse a ligandos de las plaquetas (GpIb/IX y GpIIb/IIIa), en el subendotelio (colágeno) y en el torrente sanguíneo (FVIII)7. La proteína lleva a cabo una incorporación postraduccional, sus secuencias señal son reconocidas por el complejo Sec62/63m que está asociado con el translocón en la membrana del retículo endoplásmico. El propéptido de 2791aa inicia el proceso de formación de la proteína del VWF y forma dímeros mediante puentes disulfuro en las posiciones carboxiterminales, por la enzima disulfuroisomerasa. Posteriormente se lleva a cabo la glucosilación y se transporta al aparato de Golgi donde la proteína madura de 2051aa forma puentes de disulfuro en las porciones aminoterminales de los dímeros, y por consiguiente, series de multímeros de diferentes tamaños que van desde una sola unidad fundamental de 225kDa hasta 120,000kDa (figs. 1 y 2)11,12.
, donde termina su maduración formando multímeros. Una pequeña cantidad de VWF inmaduro, dímeros y pequeños multímeros se liberan de manera constitutiva.")
Esquema del procesamiento y secreción del factor de von Willebrand en células endoteliales. El VWF se forma en el retículo endoplásmico, donde se dimeriza glucosila. Los dímeros se trasportan al aparato de Golgi y continúan en los gránulos de secreción (cuerpos de Weibel-Palade), donde termina su maduración formando multímeros. Una pequeña cantidad de VWF inmaduro, dímeros y pequeños multímeros se liberan de manera constitutiva.
Hay 2 vías involucradas en la secreción del VWF. La vía constitutiva se relaciona con la síntesis del VWF plasmático de origen endotelial que se almacena en los cuerpos de Weibel-Palade. Y la vía regulada, que involucra la liberación del VWF totalmente multimerizado, almacenado en los gránulos alfa, en megacariocitos y plaquetas (fig. 2)13,14.
El VWF comparte estos sitios de almacenamiento con otras proteínas que se liberan en respuesta a una variedad de estímulos fisiológicos y farmacológicos, incluyendo trombina, fuerza de cizallamiento (shear stress) y desmopresina15. Actualmente se sabe que luego de su liberación de la célula donde se sintetiza, los multímeros del VWF extremadamente grandes y de alto peso molecular se unen a la superficie de la célula endotelial mediante interacción con la proteína P selectina de los cuerpos de Weibel-Palade. En este sitio, los multímeros del VWF se ven sometidos a la fuerza de cizallamiento del flujo sanguíneo, y a reducción fisiológica del tamaño de los multímeros por fragmentación proteolítica controlada, por la metaloproteinasa ADMTS-13 que actúa sobre los multímeros del VWF en el dominio A2, entre los aa 1605 y 1606. Este proceso origina las diferentes formas del factor que van desde dímeros simples, hasta las 20 unidades que forman el multímero más complejo. La degradación proteolítica de los multímeros es un evento normal, ya que estos tienen un elevado potencial trombogénico, por los sitios de interacción con las plaquetas, y la pared de los vasos sanguíneos. En condiciones de homeostasis esta reacción inhibe el crecimiento del trombo que forman las plaquetas16,17.
La estructura del VWF indica que su principal papel consiste en unirse con diversos ligandos en el torrente sanguíneo y la pared del vaso dañado (fig. 3). Por lo tanto, las 3 funciones fisiológicas de la proteína son: A) Mediar la adhesión de las plaquetas a los sitios de daño vascular al unirse al receptor plaquetario GpIb/IX, y al colágeno en el subendotelio vascular. B) Facilitar la agregación plaquetaria por medio de su unión al receptor plaquetario GpIIb/IIIa; y C) Unirse al FVIII y protegerlo de la degradación proteolítica provocada por la proteína C activada en el torrente sanguíneo16. Finalmente, estos contactos alcanzan un umbral que señala el evento de la activación plaquetaria. Entonces, las plaquetas se adhieren de manera estable a la pared del vaso dañado y experimentan una respuesta de agregación a través de un evento mediado por un receptor plaquetario, la GpIIb/IIIa17.

Descripción esquemática de la interacción del factor de von Willebrand y las plaquetas activadas durante la formación del tapón plaquetario en la hemostasia. Las plaquetas se adhieren de manera transitoria al factor de von Willebrand, y su función es actuar como un puente entre el receptor GpIb/IX en la superficie de las plaquetas y las fibrillas de colágeno del subendotelio.
La enfermedad de von Willebrand es el trastorno hereditario de la coagulación más común en los seres humanos, presenta una distribución mundial y también es común en otras especies animales como perros y cerdos18. Su prevalencia varía dependiendo del enfoque que se tome para definir el diagnóstico. En por lo menos 2 grandes estudios prospectivos se ha encontrado que hasta el 1% de la población predominantemente pediátrica presenta síntomas y signos de laboratorio de enfermedad de von Willebrand19,20. Se cree que la prevalencia de la enfermedad de von Willebrand con síntomas hemorrágicos es de aproximadamente 1 por 1,00021. En México no hay un registro epidemiológico de la enfermedad; solo se han informado algunos estudios desde el punto de vista clínico y hematológico, y recientemente un estudio piloto en 133 pacientes con sospecha de enfermedad de von Willebrand22.
Generalidades de la enfermedad de von WillebrandLa enfermedad de von Willebrand se transmite en forma autosómica dominante; es el desorden hemorrágico heredado más común. Es causado por la disminución en la cantidad de VWF o por la presencia de un VWF cualitativamente anormal en la circulación. De forma poco común, la enfermedad de von Willebrand puede ser un trastorno adquirido. Las manifestaciones de la enfermedad son sangrados cutáneos mucosos (epistaxis, sangrado de las encías, sangrado de otras mucosas, equimosis, sangrado en procedimientos dentales, etc.). Su diagnóstico depende por completo de las pruebas en el laboratorio de coagulación y su clasificación exige estudios especiales (como la determinación de los multímeros del VWF). Es importante clasificar el tipo ya que esto tiene importancia para elegir el tratamiento. El tratamiento en la mayoría de las veces es con desmopresina, solo en los casos más severos se reemplaza el factor23.
Diagnóstico clínicoUna historia clínica veraz, detallada y completa permitirá al médico hacer una aproximación respecto al tipo de alteración de la hemostasia que exista. Es necesario tener claro que mientras que una historia familiar positiva es de ayuda para aclarar el diagnóstico, una historia familiar negativa no excluye la posibilidad de una anomalía hemorrágica congénita. Incluso, en ocasiones la historia familiar puede no proporcionar evidencias concluyentes. La historia familiar en pacientes con enfermedad de von Willebrand muchas veces es poco clara; debe sospecharse en personas con hemorragias mucocutáneas excesivas, tales como: hematomas sin traumas reconocidos, la aparición espontánea, o con mínimos traumatismos; también la hemartrosis es siempre anormal e indica la existencia de una coagulopatía grave. Lo mismo puede decirse de los hematomas musculares espontáneos. Ambos constituyen el síntoma primordial de la hemofilia, y su aparición es muy rara en otras diátesis hemorrágicas, salvo en la enfermedad de von Willebrand severa, sobre todo la de tipo 3. Asimismo, hemorragias prolongadas de nariz recurrentes y hemorragias en cavidad bucal, incluyendo sangrado de las encías después de cepillarse los dientes o usar hilo dental o sangrado prolongado después de la limpieza dental o extracciones. Puede incluso presentarse hematuria, sangrado excesivo o prolongado después de una cirugía o trauma, y mujeres afectadas también suelen experimentar menorragia (por lo general ocurre desde la menarquia), y sangrado prolongado o excesivo después del parto. También es importante tomar en cuenta diferentes factores, algunos de los cuales aumentan la concentración del VWF: embarazo, ejercicio, trauma, cirugía, edad; y su nivel es mayor en individuos del grupo sanguíneo A, que en el O. En cambio, otros causan su disminución: hipertiroidismo, falla renal, enfermedad hepática, aterosclerosis, estados inflamatorios, cáncer, diabetes e hipotiroidismo24,25.
Pruebas de laboratorio químico hematológicoEl laboratorio químico hematológico proporciona información de gran utilidad que debe ser interpretada en relación con el contexto clínico, lo que brinda mayor posibilidad de un diagnóstico certero y un tratamiento adecuados. Las pruebas se dividen en básicas, las que nos permiten tener un panorama general acerca de la condición del enfermo, que incluyen: citometría hemática, tiempo de sangrado, tiempo de protrombina, tiempo de tromboplastina parcial activada, y tiempo de trombina; posteriormente, las pruebas que estudian la hemostasia primaria, secundaria y la fibrinólisis, así como las pruebas encaminadas al diagnóstico de problemas trombóticos26,27.
Pruebas básicasSe consideran pruebas básicas el recuento de plaquetas, el tiempo de sangrado, el tiempo de protrombina, el tiempo de tromboplastina parcial activada y el tiempo de trombina. Las 2 primeras exploran la hemostasia primaria, mientras que las 3 restantes evalúan la hemostasia secundaria. Su normalidad, salvo en raras excepciones, descarta los trastornos hemostáticos con significado clínico27,28. Su alargamiento puede ser debido a deficiencia de un factor o a la existencia de un anticuerpo, inhibidor de proteínas de la coagulación. La corrección, o no, de la prueba, al mezclar plasma del paciente con plasma normal, informará si se trata de una deficiencia o de un inhibidor29.
Pruebas específicasPermiten cuantificar factores individuales. Se utilizan cuando una o más pruebas básicas aparecen alteradas, o aun siendo normales, cuando existe sospecha clínica de una coagulopatía sistémica. Son de 2 tipos: ensayo, funcionales e inmunológicos. Los primeros, a su vez, pueden ser métodos coagulométricos que utilizan plasmas deficientes del factor a analizar.
Los métodos inmunológicos, mediante anticuerpos, valoran la concentración del antígeno de la proteína (pero no su función). La discrepancia entre los niveles obtenidos por métodos funcionales e inmunológicos revela la existencia de anomalías moleculares.
Dentro de las pruebas específicas se encuentran la actividad del VWF como cofactor de la ristocetina (VWF:RCo), que depende de la presencia de los multímeros grandes, VWF antigénico (VWF:Ag), FVIII coagulante (FVIII:C), aglutinación de plasma rico en plaquetas con ristocetina (RIPA por sus siglas en inglés); la mayoría de los subtipos tienen una disminución en la respuesta a la ristocetina, este es un agonista del receptor de la GpIb-IX y el VWF. Además, se pueden incluir la capacidad de unión del VWF al colágeno y al FVIII:C (VWF:CB y VWF:VIII) y el análisis de función plaquetaria (PFA-100®). Este último podría sustituir en un futuro al tiempo de sangrado, cuyo empleo es controvertido.
Análisis de los multímeros del factor de von WillebrandEl análisis de los multímeros del VWF, en función de su peso molecular, se hace mediante electroforesis. Los multímeros son vistos mediante autorradiografía después de la incubación con anticuerpo contra el VWF marcados con inmunoperoxidasa o fosfatasa alcalina. Esta técnica permite el diagnóstico con certeza de los diferentes tipos de la enfermedad de von Willebrand según su patrón de multímeros y tiene lugar solo en los laboratorios de referencia24,25,30,31.
Clasificación de la enfermedad de von WillebrandEn 2006 fue la última vez que la Sociedad Internacional de Trombosis y Hemostasia (International Society of Thrombosis and Haemostasis, ISTH) publicó sus recomendaciones respecto a la clasificación de la enfermedad de von Willebrand, que permiten que dicha enfermedad se subdivida como deficiencias cuantitativas (tipos 1 y 3) o cualitativa (tipo 2), basado principalmente en el fenotipo de la proteína del VWF25,32.
Enfermedad de von Willebrand tipo 1Esta es la forma más común de la enfermedad de von Willebrand y representa cerca del 80% de todos los casos. Su transmisión es autosómica dominante con penetrancia incompleta. Se caracteriza por una reducción de leve a moderada (0.45-0.05U/ml) en los niveles plasmáticos de VWF: Ag y VWF:RCo. El VWF es normal desde el punto de vista funcional, al igual que el rango de multímeros de VWF plasmático, mientras que el nivel plasmático de FVIII:C se reduce en proporción al nivel de VWF. Estos pacientes manifiestan un espectro de síntomas hemorrágicos mucocutáneos cuya gravedad, por lo general, se correlaciona con el nivel de deficiencia del VWF. Los estudios moleculares indican que además de las mutaciones en el gen del VWF y del grupo sanguíneo ABO, alteraciones en otros genes pueden influir en la disminución leve del VWF, como los involucrados en la regulación de los niveles plasmáticos del VWF y del FVIII33. Si bien parece haber muchas mutaciones diferentes causantes de la enfermedad tipo 1, por lo menos una, la mutación que origina la sustitución de tirosina por cisteína en el codón 1584, se encuentra en el 10-20% de los pacientes en EE. UU. y Europa25,32,34.
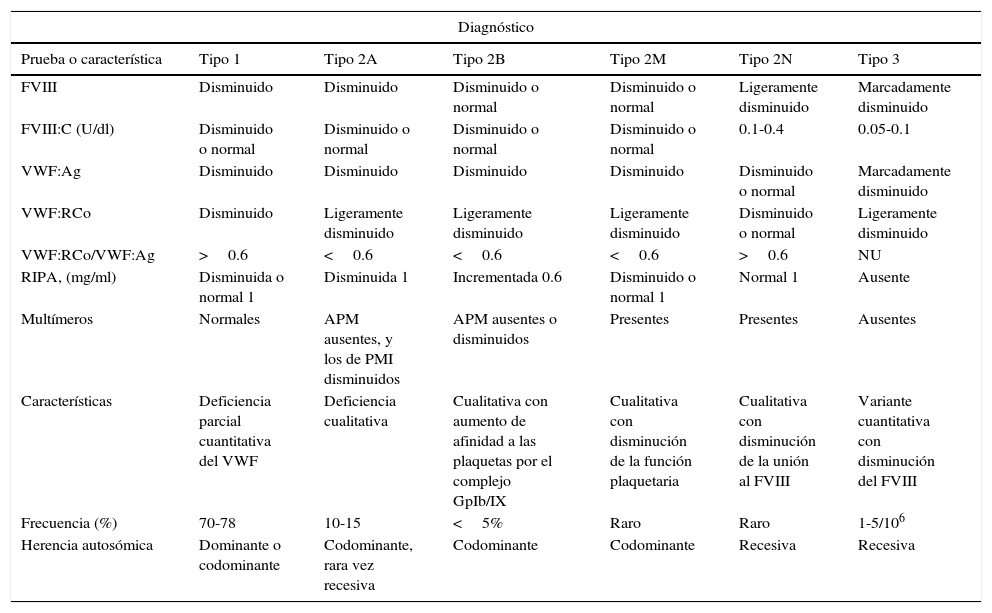
Resumen para algunos resultados del tipo 1. A continuación se resumen algunos de los resultados de laboratorio esperados para el tipo 1: FVIII, VWF:Ag y VWF:RCo disminuidos; FVIII:C y ristocetina (RIPA) disminuidos o normales. VWF:RCo/VWF:Ag>0.6. Multímeros normales, pero con deficiencia parcial cuantitativa. Frecuencia: 70-80% y herencia autosómica dominante o co-dominante25,32–34.
Enfermedad de von Willebrand tipo 3La enfermedad de von Willebrand tipo 3 tiene una prevalencia de entre 1-3 por millón en la mayoría de las poblaciones, aunque en algunos lugares donde los matrimonios consanguíneos son frecuentes la prevalencia es considerablemente mayor. El trastorno es autosómico recesivo y la mayoría de los padres de pacientes con enfermedad tipo 3 muestran pocos, si no es que nulos, síntomas hemorrágicos; sin embargo, en los individuos afectados es la forma más severa de la enfermedad.
En la enfermedad tipo 3, los niveles de VWF:Ag y de VWF:RCo siempre son<0.05U/ml, y con frecuencia indetectables. El nivel plasmático de FVIII:C se reduce a entre 0.01 y 0.10U/ml. Por lo general no hay multímeros plasmáticos. Estos pacientes manifiestan hemorragias mucocutáneas recurrentes graves, así como hemorragias frecuentes musculoesqueléticas, y en tejidos blandos. Con el transcurso del tiempo, si el tratamiento no es adecuado, se presenta daño musculoesquelético crónico y los pacientes de edad mediana podrían requerir cirugía de reemplazo articular25,32–34.
Resumen de resultados de laboratorio esperados para el tipo 3. FVIII, VWF:Ag y FVIII:C marcadamente disminuidos. VWF:RCo ligeramente disminuido. ristocetina (RIPA) y multímeros ausentes. Variante cuantitativa con disminución del FVIII. Frecuencia: 1-5/106 y herencia autosómica recesiva.
Enfermedad de von Willebrand tipo 2La actual clasificación de la enfermedad de von Willebrand reconoce 4 formas cualitativas distintas del padecimiento: los subtipos 2A, 2B, 2M, y 2N25,32–34.
Enfermedad de von Willebrand tipo 2A
Este padecimiento se caracteriza por pérdida de la función del VWF dependiente de las plaquetas debido a la ausencia de formas con alto peso molecular de la proteína. Existe ya sea por una incapacidad biosintética para producir estos multímeros o porque estos producidos, segregados y subsiguientemente degradados de manera prematura en el plasma. La característica típica de la enfermedad tipo 2A es una relación entre el VWF:RCo y el VWF:Ag (<0.6), con ausencia de multímeros de VWF de alto peso molecular y afectación de la capacidad de aglutinación plaquetaria inducida por la RIPA. Las mutaciones sustitutivas que causan la enfermedad tipo 2A se han ubicado en los dominios D2, D3, A1, A2 y C terminal del gen del VWF25,32–34.
Resumen para algunos resultados del tipo 2A. FVIII, VWF:Ag, VWF:RCo y RIPA disminuidos. FVIII:C disminuido o normal. VWF:RCo/VWF:Ag>0.6. Multímeros de alto peso molecular ausentes, y los de peso molecular intermedio, disminuidos (deficiencia cualitativa). Frecuencia: 10-15% y herencia codominante, rara vez recesiva.
Enfermedad de von Willebrand tipo 2B.
Este subtipo de la enfermedad de von Willebrand representa un clásico rasgo genético de ganancia de función. El trastorno es el resultado de una variedad de mutaciones sustitutivas dominantes en la región de unión de la GpIb en el dominio A1 del VWF. Estas mutaciones incrementan la capacidad de adherencia del VWF a este receptor plaquetario y producen interacciones espontáneas entre VWF y las plaquetas en el torrente sanguíneo, un fenómeno que no ocurre con el VWF normal. En las muestras de sangre, esta interacción puede observarse como acumulación de las plaquetas y esta misma anormalidad con frecuencia produce trombocitopenia crónica leve. En otras pruebas, la relación entre VWF:RCo y VWF:Ag a menudo será<0.6, con un déficit de multímeros de VWF de alto peso molecular en el plasma porque estos se han ligado a las plaquetas. Otra prueba importante para confirmar la presencia del tipo 2B de la enfermedad es la demostración del incremento en la capacidad de aglutinación plaquetaria inducida por la RIPA; se detecta por la agregación plaquetaria a bajas concentraciones de ristocetina<0.6mg/ml.
Este conjunto de hallazgos clínicos y de laboratorio también pueden observarse en un trastorno plaquetario hereditario poco común, la seudoenfermedad de von Willebrand o enfermedad de von Willebrand tipo plaquetario. En este rasgo hereditario dominante, las mutaciones sustitutivas de ganancia de función se encuentran en el gen de la GP plaquetaria Ib y causan incremento en la afinidad de adhesión al dominio A1 del VWF. Para diferenciar el tipo 2B de la enfermedad de von Willebrand del tipo plaquetario de la misma se necesitan pruebas de aglutinación inducida por ristocetina a plaquetas lavadas del paciente y mezcladas con plasma normal (que mostrará una mayor reactividad en el caso de enfermedad de von Willebrand tipo plaquetario, pero no en el tipo 2B), o realizar el análisis de los genes del VWF y de la GpIb25,32–34.
Resultados para el tipo 2B. FVIII y FVIII:C disminuidos o normales. VWF:Ag ligeramente disminuido. VWF:RCo disminuido. RCo/VWF:Ag<0.6. RIPA incrementada. Multímeros de alto peso molecular ausentes o disminuidos, se describe como una variante cualitativa con aumento de afinidad a las plaquetas por el complejo GpIb/IX, una frecuencia<5% y una herencia codominante.
Enfermedad de von Willebrand tipo 2M.
Este subtipo se caracteriza por la pérdida de función, equivalente al tipo 2B de la enfermedad. La mayoría de las mutaciones sustitutivas que causan el tipo 2M de la enfermedad de von Willebrand también han sido localizadas en el domino A1 del VWF. En la enfermedad tipo 2M, la relación entre el VWF:RCo y el VWF:Ag también es<0.6, pero las características que la diferencian del tipo 2B incluyen la ausencia de acumulación de plaquetas (y por ende de trombocitopenia) y la presencia de un patrón normal de multímeros en el plasma25,32–34.
Resultados para el tipo 2M. FVIII y FVIII:C disminuidos o normales. VWF:Ag ligeramente disminuido. VWF:RCo disminuido. RCo/VWF:Ag<0.6. RIPA disminuida o normal. Multímeros presentes, se trata de una variante cualitativa con disminución de la función plaquetaria, raramente se presenta y con una herencia codominante.
Enfermedad de von Willebrand tipo 2N.
Esta última forma mutante cualitativa de la enfermedad de von Willebrand es diferente de todas las anteriores en varios aspectos. Su patrón de herencia es autosómico recesivo y frecuentemente la única anormalidad de laboratorio es un nivel plasmático reducido de FVIII (por lo general entre 0.10 y 0.40U/ml). La enfermedad de von Willebrand tipo 2N constituye uno de los diagnósticos diferenciales de un nivel aislado, de leve a moderadamente bajo de FVIII33–37.
Los pacientes pueden ser homocigotos para mutaciones de sustitución o heterocigotos compuestos para 2 mutaciones diferentes. También pueden tener mutaciones en el sitio de unión o una mutación nula. Cerca de 20 mutaciones han sido descritas. La mayoría de ellas se localizan entre los exones 18 y 20, que afectan el dominio de unión al FVIII. Se han descrito otras mutaciones en los exones 17 y del 21 al 27 que están fuera del sitio de unión al FVIII, y también son responsables de la disminución de la capacidad de unión entre VWF y FVIII. La mutación R854Q es la más frecuentemente reportada.
Resultados para el tipo 2N. FVIII y FVIII:C disminuidos. VWF:Ag ligeramente disminuido o normal. VWF:RCo y RIPA normales. Multímeros presentes. Es una variante cualitativa con disminución de la unión al FVIII, muy rara y con una herencia autosómica recesiva (tabla 1).
Pruebas de laboratorio químico hematológico y herencia de los tipos de la enfermedad de von Willebrand
| Diagnóstico | ||||||
|---|---|---|---|---|---|---|
| Prueba o característica | Tipo 1 | Tipo 2A | Tipo 2B | Tipo 2M | Tipo 2N | Tipo 3 |
| FVIII | Disminuido | Disminuido | Disminuido o normal | Disminuido o normal | Ligeramente disminuido | Marcadamente disminuido |
| FVIII:C (U/dl) | Disminuido o normal | Disminuido o normal | Disminuido o normal | Disminuido o normal | 0.1-0.4 | 0.05-0.1 |
| VWF:Ag | Disminuido | Disminuido | Disminuido | Disminuido | Disminuido o normal | Marcadamente disminuido |
| VWF:RCo | Disminuido | Ligeramente disminuido | Ligeramente disminuido | Ligeramente disminuido | Disminuido o normal | Ligeramente disminuido |
| VWF:RCo/VWF:Ag | >0.6 | <0.6 | <0.6 | <0.6 | >0.6 | NU |
| RIPA, (mg/ml) | Disminuida o normal 1 | Disminuida 1 | Incrementada 0.6 | Disminuido o normal 1 | Normal 1 | Ausente |
| Multímeros | Normales | APM ausentes, y los de PMI disminuidos | APM ausentes o disminuidos | Presentes | Presentes | Ausentes |
| Características | Deficiencia parcial cuantitativa del VWF | Deficiencia cualitativa | Cualitativa con aumento de afinidad a las plaquetas por el complejo GpIb/IX | Cualitativa con disminución de la función plaquetaria | Cualitativa con disminución de la unión al FVIII | Variante cuantitativa con disminución del FVIII |
| Frecuencia (%) | 70-78 | 10-15 | <5% | Raro | Raro | 1-5/106 |
| Herencia autosómica | Dominante o codominante | Codominante, rara vez recesiva | Codominante | Codominante | Recesiva | Recesiva |
APM: multímeros de alto peso molecular; FVIII: factor VIII de la coagulación; FVIII:C: factor VIII coagulante; NU: no es útil; PMI: multímeros de peso molecular intermedio; RIPA: agregación plaquetaria inducida por ristocetina; VWF: factor de von Willebrand; VWF:Ag: VWF antigénico; VWF:RCo: VWF como cofactor de la ristocetina.
Las variaciones alélicas en el genoma del VWF implican efectos sobre el fenotipo, perjudiciales, neutrales o benéficos. Las variantes normales son muy comunes, y se han descrito alrededor de 150 hasta la fecha, como la variante c.1451A>G que provoca un cambio de una histidina por una arginina en la posición 484 del exón 13, las cuales están relacionadas con las diferentes poblaciones estudiadas38,39.
Existe una gran variedad de factores implicados en el origen de variaciones patológicas, debidas a cambios en un solo nucleótido, principalmente en el dominio D3 (fig. 1), como la variante «Vicenza» (c.3614G>A) en el exón 27, que provoca un cambio de una arginina por una histidina en la posición 1205 del VWF y con ello disminuye el tiempo en que el VWF permanece en el plasma; esta mutación es una de las más estudiadas y se relaciona con la enfermedad de von Willebrand tipo 140,41. Incluso existe un estudio molecular del gen VWF en pacientes mestizos mexicanos, en el que se describen 3 nuevas mutaciones: E1447Q en un paciente con la enfermedad de von Willebrand tipo 1 y diabetes; P2781S en un paciente con tipo 2M; y P812L en otro paciente tipo 1/2N22,42. Este alto grado de polimorfismos en el VWF, junto con el gran tamaño del gen y la presencia de un seudogén parcial, hacen que la secuenciación completa del gen y la interpretación de datos sean difíciles y complejas. Estas variaciones y sus frecuencias pueden ser consultadas ampliamente en la base de datos del International Society on Thrombosis and Haemostasis-Scientific and Standardization Committee ISTH-SSC VWF Online (VWFdb).
Como ha sido descrito, la gran cantidad de variaciones que se presentan sobre el gen, y con ellas sus efectos sobre la estructura y función del VWF, causan diferentes formas de enfermedad de von Willebrand. Además del hecho de que otras enfermedades pueden estar relacionadas con defectos cuantitativos y cualitativos en el VWF. Las nuevas evidencias proporcionan información precisa sobre los mecanismos de la enfermedad y el riesgo de sangrado asociado con la deficiencia o anormalidad del VWF43.
TratamientoEl tratamiento depende del subtipo de la enfermedad de von Willebrand, sin embargo las terapias para prevenir o controlar el sangrado recomiendan: aumentar la concentración plasmática de VWF por liberación endógena a través de la estimulación de células endoteliales con desmopresina. Asimismo, la terapia transfusional con productos sanguíneos. Así como utilizar agentes que promuevan la hemostasia y la cicatrización de heridas, pero que no alteran sustancialmente la concentración plasmática de VWF.
Las decisiones terapéuticas dependen del tipo y la gravedad de la enfermedad de von Willebrand, así como de la gravedad del sangrado, y su naturaleza real o potencial. El tratamiento es variable y con frecuencia se basa en la experiencia local y la preferencia del médico. Existen recomendaciones estándar para guiar el tratamiento de la enfermedad de von Willebrand44,45.
DiscusiónLa biología molecular ha permitido la caracterización del gen del VWF, así como la determinación de variantes alélicas y mutaciones. Una de las aplicaciones más importante de la biología molecular está dirigida al diagnóstico molecular, el cual se apoya no solo en pruebas básicas o de escrutinio de laboratorio químico clínico, las pruebas especiales y en la genética. Las pruebas de biología molecular tienen un papel cada vez más importante en el diagnóstico de la enfermedad de von Willebrand, mejorando en gran medida la capacidad para caracterizar las variantes genéticas de la enfermedad. Así como en la investigación de alteraciones en otros genes que pueden estar involucrados en la regulación de la síntesis, procesamiento, secreción y el control de los niveles plasmáticos del VWF. Del mismo modo, dada la gravedad del fenotipo de la enfermedad de von Willebrand tipo 3, el diagnóstico prenatal genético de esta variante ofrece resultados a las familias y sus médicos para tomar decisiones informadas sobre la planificación familiar. Sin embargo, dada la complejidad que implica el estudio completo del gen es difícil realizar la búsqueda de alguna mutación en particular, por lo que aún no se han integrado las estrategias de biología molecular entre las pruebas de diagnóstico disponibles. Por otro lado, el análisis de los multímeros del VWF es una metodología que cumple con las características para el diagnóstico, sin embargo, es técnicamente exigente y no es fácil de estandarizar. Tomando en consideración que aún en los centros de tercer nivel en nuestro país los enfermos de von Willebrand no cuentan con un diagnóstico definitivo, es necesario implementar estas metodologías para su estudio y mejorar su diagnóstico.
ConclusionesLa enfermedad de von Willebrand es muy heterogénea debido a los mecanismos moleculares que producen los distintos fenotipos clínicos y de laboratorio. En México existen pocos trabajos relacionados con esta enfermedad, por ello es fundamental realizar un estudio integral que incluya aspectos clínicos, pruebas de laboratorio básicas y especiales en este tipo de pacientes de nuestra población, para establecer un diagnóstico correcto y desarrollar nuevos enfoques terapéuticos para su tratamiento, que permita prevenir y/o corregir las alteraciones que presentan, y así poder ofrecer atención médica y asesoramiento genético adecuados.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.