





Los aneurismas de la aorta torácica (AAT) son una patología relativamente frecuente, responsable de hasta 15.000 muertes anuales en Estados Unidos, lo que los sitúa como la 13.a causa principal de muerte en dicho país1,2. Se detecta un sustrato genético predisponente hasta en el 20% de los pacientes diagnosticados de esta patología1,3–5.
Los AAT familiares pueden diferenciarse en sindrómicos (los que aparecen acompañados de manifestaciones fenotípicas a otros niveles) y no sindrómicos (los que aparecen como manifestación aislada, pero con agregación familiar, sugiriendo una base genética).
El máximo exponente del primer grupo es el síndrome de Marfan (SMF), causado por una mutación en el gen de la fibrilina-1 (FBN1). Además de un fenotipo característico, los pacientes portadores de las mutaciones genéticas responsables del SMF desarrollan AAT muy agresivos, de rápido crecimiento y con alto riesgo de rotura y disección a edades tempranas de la vida (mediana de edad, 24,8 años).
Se debe establecer el diagnóstico diferencial entre el SMF y múltiples patologías del tejido conectivo con características fenotípicas y manifestaciones clínicas similares3. La mayoría de estas enfermedades son monogénicas y tienen un patrón de herencia autosómico dominante, y en ocasiones el diagnóstico diferencial se convierte en un auténtico reto (tabla 1).
Diagnóstico diferencial del síndrome de Marfan
| Aneurismas de la aorta torácica sindrómicos | No fibrilinopatías |
| • Síndrome de Loeys-Dietz | |
| • Síndrome de Ehler-Danlos tipo IV | |
| • Síndrome de Turner | |
| • Síndrome de Beals | |
| • Síndrome de Noonan | |
| • Síndrome de Alagille | |
| • Enfermedad poliquística renal autosómica dominante | |
| Fibrilinopatías | |
| • Síndrome de Shprintzen-Goldberg | |
| • Síndrome de Weill-Marchesani | |
| • Fenotipo MASS | |
| Aneurismas de la aorta torácica familiares no sindrómicos | TAAD1, TAAD2, TAAD3, TAAD4, TAAD5, FAA1 y TAAD-asociado a ductus arterioso persistente |
| Válvula aórtica bicúspide | |
Entre los síndromes genéticos que pueden acompañarse de AAT destacan:
Fenotipo MASS (mitral valve-aorta-skeleton-skin)3El término se acuñó para denominar a los pacientes con un trastorno sistémico del tejido conectivo que no cumplían los criterios diagnósticos de otros síndromes conocidos, especialmente del SMF, con el que comparte muchas de sus manifestaciones fenotípicas. Se incluye, junto con otras entidades, dentro del grupo de las fibrilinopatías (enfermedades producidas por mutaciones en el gen de la fibrilina) y se caracteriza por la presencia de miopía, prolapso de la válvula mitral, dilatación aórtica (leve y no progresiva) y anormalidades en el tejido cutáneo y musculoesquelético. Al menos dos sistemas deben estar afectos para establecer el diagnóstico.
Síndrome de Loeys-Dietz3–6Es una enfermedad autosómica dominante resultado de una mutación en alguno de los genes que codifican para el receptor del factor de crecimiento transformante beta (TGFBR1 o TGFBR2). Se pueden distinguir dos variantes fenotípicas (tabla 2).
Síndrome de Loeys-Dietz
| Loeys-Dietz tipo I | Loeys-Dietz tipo II | |
| Fenotipo | Hipertelorismo. CraneosinostosisÚvula bífida/paladar hendidoTortuosidad arterial y aneurismas | Sin otras anomalías craneofaciales, excepto úvula bífidaSimilar al síndrome de Ehlers-Danlos tipo IV |
| Genes mutados | TGFBR1 y TGFBR2 | TGFBR1 y TGFBR2 |
| Prevalencia | Desconocida | Desconocida |
| Pronóstico | Mediana de supervivencia, 37 años. Fallecimiento a la edad media de 26 años | Mediana de supervivencia, 37 años. Fallecimiento a la edad media de 26 años |
Los aneurismas de aorta son muy frecuentes (98%), aparecen a edades tempranas y se caracterizan por un alto riesgo de disección y/o rotura, incluso con diámetros <5cm4,6. Además, hasta el 53% de los pacientes desarrollan aneurismas en otras localizaciones. En general, los pacientes con manifestaciones craneofaciales más severas presentan una enfermedad arterial más agresiva.
Recomendaciones en el síndrome de Loeys-Dietz4- •
Estudio completo de imagen para evaluar la aorta en el momento del diagnóstico y cada 6 meses para ver el ritmo de crecimiento del AAT.
- •
Resonancia magnética (RM) anual craneo-torácico-abdomino-pélvica para detectar posibles aneurismas vasculares sistémicos.
- •
Reparación quirúrgica de los AAT cuando el diámetro interno supere los 4,2cm en el ecocardiograma transesofágico o el diámetro externo sea mayor de 4,5cm en la tomografía computarizada (TC) o en la RM.
Es producido por una mutación en el gen que codifica para el colágeno tipo 3 (COL3A1) y se transmite de forma autosómica dominante. Se caracteriza por una fragilidad vascular y visceral extrema, que puede conducir a roturas vasculares y viscerales espontáneas o ante mínimos traumatismos. Otras manifestaciones de la enfermedad, como la hiperlaxitud cutánea y articular, son menos marcadas que en los otros subtipos. La mayor parte de las muertes se producen por roturas vasculares.
Recomendaciones en estos pacientes7Se recomienda realizar pruebas de imagen no invasivas por el alto riesgo de rotura vascular. Se desconoce la utilidad de la cirugía aórtica para reparar los AAT no complicados. En caso de rotura o disección aórtica sí está indicada la cirugía urgente, prestando especial cuidado a las anastomosis vasculares por la tendencia a la hemorragia, la fragilidad vascular y las dificultades para la cicatrización que conllevan este síndrome.
Síndrome de Turner4,5Síndrome causado por una monosomía del cromosoma X (cariotipo 45,X0). Las pacientes afectadas muestran un fenotipo característico (estatura baja, cuello corto, infertilidad) que puede incorporar distintas manifestaciones cardiovasculares, como coartación de la aorta, cardiopatía isquémica precoz, válvula aórtica bicúspide y AAT (hasta en el 40%). La incidencia de disección de la aorta torácica (DAT) en estas pacientes es claramente superior a la de la población general (×6), con una mediana de edad de 31 años, pero aun así sigue siendo claramente inferior a la incidencia de AAT o DAT en los pacientes con las otras enfermedades sindrómicas ya mencionadas.
Recomendaciones en pacientes con síndrome de Turner4Estas pacientes deben realizarse una prueba de imagen inicial para descartar válvula aórtica bicúspide, coartación de aorta o AAT.
Si la prueba resulta normal y no hay factores de riesgo para disección aórtica, es suficiente un seguimiento cada 5-10 años con una prueba de imagen. En caso contrario se deben realizar controles anuales.
Las pacientes con síndrome de Turner gestantes o que planean un embarazo deben realizarse una prueba de imagen para determinar el riesgo de DAT.
Enfermedad poliquística renal autosómica dominante4,5Es secundaria a una mutación en los genes PKD1 y PKD2. A pesar de que la complicación más frecuente son las hemorragias subaracnoideas secundarias a la rotura de aneurismas cerebrales, se relaciona también con un aumento en la incidencia de AAT y disecciones aórticas tipo A.
Síndrome de Beals, o aracnodactilia contractural congénita3,8Es debido a mutaciones en el gen de la fibrilina 2 (FBN2), heredadas de modo autosómico dominante. A pesar de que el fenotipo puede ser similar al del SMF, se diferencia por la aparición de contracturas de flexión múltiples, aracnodactilia, cifoescoliosis grave, pabellones auriculares anormales e hipoplasia muscular. Se asocia a elongación aórtica y AAT, pero no se ha documentado progresión hacia la DAT.
Otros síndromes asociados a AAT3,4Síndrome de Noonan y síndrome de Alagille.
Aneurismas de la aorta torácica familiares no sindrómicos (FTAAD)La mayoría de los AAT y disecciones aórticas familiares se producen en pacientes que no pueden encuadrarse en ninguno de los síndromes ya descritos. Los estudios de agregación familiar sugieren que entre el 11 y el 19% de los pacientes con AAT o disecciones presentan un familiar de primer grado con dicho antecedente3–5.
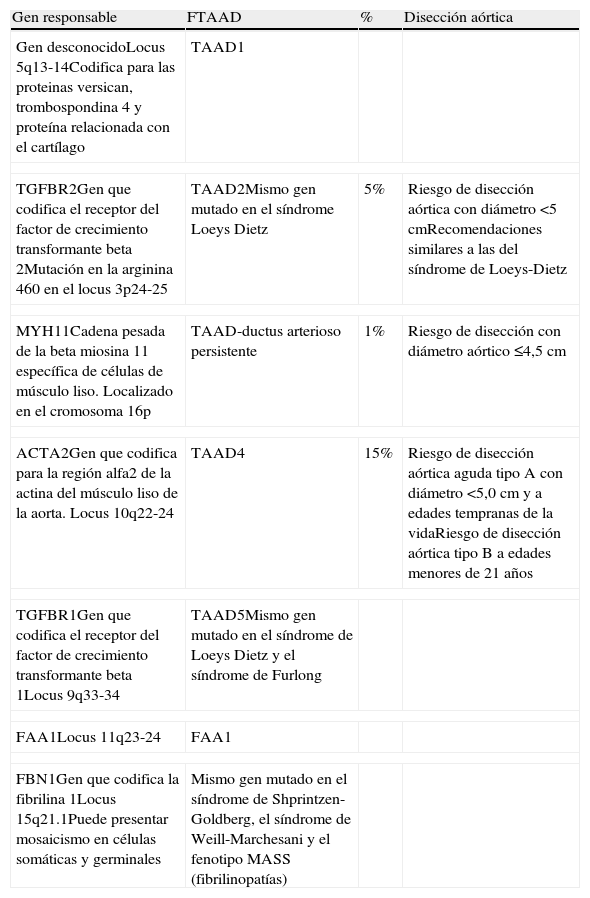
En general, en estos pacientes las complicaciones (rotura o disección) tienden a producirse a edades más tempranas (mediana de edad de 56,8 años) en comparación con los pacientes con aneurismas esporádicos (64,3 años), aunque sin alcanzar la precocidad de los AAT sindrómicos5. La dilatación de la aorta puede afectar tanto a la porción tubular de la aorta ascendente como a los senos de Valsalva1. Tanto la edad de aparición de la clínica como la velocidad de progresión de las lesiones son muy variables, incluso dentro de los componentes de una misma familia. Desde el punto de vista genético, los FTAAD se caracterizan por una marcada heterogeneidad, habiéndose identificado hasta el momento 7 loci diferentes: TAAD1, TAAD2, TAAD3, TAAD4, TAAD5, FAA1 y TAAD-asociado a ductus arterioso persistente3 (tabla 3). El patrón de herencia es autosómico dominante con penetrancia incompleta (menor en el sexo femenino)1,3,5. Esta heterogeneidad genética es un claro inconveniente para su diagnóstico y, a día de hoy, los genes ya descubiertos explican sólo el 20% del total de los FTAAD.
Aneurismas familiares de la aorta torácica no sindrómicos
| Gen responsable | FTAAD | % | Disección aórtica |
| Gen desconocidoLocus 5q13-14Codifica para las proteinas versican, trombospondina 4 y proteína relacionada con el cartílago | TAAD1 | ||
| TGFBR2Gen que codifica el receptor del factor de crecimiento transformante beta 2Mutación en la arginina 460 en el locus 3p24-25 | TAAD2Mismo gen mutado en el síndrome Loeys Dietz | 5% | Riesgo de disección aórtica con diámetro <5 cmRecomendaciones similares a las del síndrome de Loeys-Dietz |
| MYH11Cadena pesada de la beta miosina 11 específica de células de músculo liso. Localizado en el cromosoma 16p | TAAD-ductus arterioso persistente | 1% | Riesgo de disección con diámetro aórtico ≤4,5 cm |
| ACTA2Gen que codifica para la región alfa2 de la actina del músculo liso de la aorta. Locus 10q22-24 | TAAD4 | 15% | Riesgo de disección aórtica aguda tipo A con diámetro <5,0cm y a edades tempranas de la vidaRiesgo de disección aórtica tipo B a edades menores de 21 años |
| TGFBR1Gen que codifica el receptor del factor de crecimiento transformante beta 1Locus 9q33-34 | TAAD5Mismo gen mutado en el síndrome de Loeys Dietz y el síndrome de Furlong | ||
| FAA1Locus 11q23-24 | FAA1 | ||
| FBN1Gen que codifica la fibrilina 1Locus 15q21.1Puede presentar mosaicismo en células somáticas y germinales | Mismo gen mutado en el síndrome de Shprintzen-Goldberg, el síndrome de Weill-Marchesani y el fenotipo MASS (fibrilinopatías) | ||
- •
Consejo genético individualizado a los familiares. Análisis genético en los familiares de primer grado, si se conoce la mutación en el caso índice.
- •
En familiares de primer grado con estudio genético negativo, se aconseja una única prueba de imagen como cribado para descartar patología aórtica.
- •
Si presentan una de las mutaciones genéticas descritas deben someterse a pruebas de imagen periódicas (cada 2 años, aproximadamente).
- •
Si uno de los parientes de primer grado del probando presenta también la mutación genética, el cribado con prueba de imagen debería ampliarse a los familiares con parentesco de segundo grado, valorando en cada caso el riesgo-beneficio.
El máximo exponente de los AAT hereditarios es el SMF. Sin embargo, existe un amplio grupo de patologías hereditarias con manifestaciones clínicas similares a las del SMF con las que es mandatorio realizar un diagnóstico diferencial. Sólo así podemos ofrecer a nuestros pacientes un tratamiento óptimo, acorde con las recomendaciones de las últimas guías de práctica clínica.
Para ello es preciso conocer los rasgos fenotípicos característicos de estas entidades, las particularidades que presentan en su historia natural y, en especial, las alteraciones genéticas específicas para cada una de ellas.
Es necesario seguir avanzando en el conocimiento de estas patologías. El futuro parece estar ligado a los progresos que se obtengan en el campo de la genética.

