






El síndrome de hipoventilación alveolar central congénita (SHACC) es un raro trastorno respiratorio del dormir, aunque cada vez más frecuentemente diagnosticado en clínicas de sueño y servicios de neumología pediátrica. Si bien se desconoce su epidemiología, en la literatura médica existen cerca de 300 casos reportados, y su incidencia es de 1 caso por cada 200,000 recién nacidos vivos. Se caracteriza por hipoventilación alveolar que se presenta o empeora durante el sueño. Es secundario a la disminución/ausencia de la respuesta ventilatoria a la hipercapnia o hipoxemia, y en el 90% de los casos es debido a una mutación tipo PARM del gen PHOX2B. Su tratamiento incluye ventilación mecánica y marcapasos diafragmático. Si la terapéutica no se inicia en forma temprana, el paciente desarrollará insuficiencia respiratoria crónica, hipertensión arterial pulmonar, cor pulmonale y la muerte.
Casos clínicosSe presentan tres casos de SHACC diagnosticados, tratados y en seguimiento en la Clínica de Trastornos Respiratorios del Dormir del Instituto Nacional de Enfermedades Respiratorias.
ConclusionesEl diagnóstico temprano es importante para el inicio del soporte ventilatorio, y para prevenir el desarrollo de complicaciones y reducir la mortalidad.
Congenital central alveolar hypoventilation syndrome (CCAHS) is a rare sleep-related breathing disorder. Although increasingly frequently diagnosed in sleep clinics and pediatric pulmonology services, its epidemiology is not known. There are about 300 reported cases reported in the literature with an incidence of 1 case per 200,000 live births. CCAHS is characterized by alveolar hypoventilation that occurs or worsens during sleep and is secondary to a reduction/absence of the ventilatory response to hypercapnia and/or hypoxemia. In 90% of the cases it is due to a PARM-type mutation of the PHOX2B gene. Treatment includes mechanical ventilation and diaphragmatic pacemaker. If therapy is not initiated promptly the patient can evolve to chronic respiratory failure, pulmonary hypertension, cor pulmonale and death.
Case reportsIn this paper we present three cases of CCAHS diagnosed, treated and followed up at the Sleep Disorders Clinic of the National Institute of Respiratory Diseases in Mexico.
ConclusionsEarly diagnosis is important to initiate ventilatory support so as to prevent any complications and to reduce mortality.
El síndrome de hipoventilación alveolar central congénita (SHACC) es un padecimiento raro caracterizado por hipoventilación alveolar que se presenta o empeora durante el sueño. Esta hipoventilación es secundaria a la disminución o ausencia de respuesta ventilatoria a la hipercapnia o hipoxemia. Por definición, no debe ser consecuencia de enfermedades específicas del sistema nervioso central, neuromusculares, metabólicas, pulmonares, cardiacas u otras lesiones que expliquen la hipercapnia1. El espectro de la enfermedad varía, y aunque la mayoría de los casos se diagnostican en el periodo neonatal, se han descrito casos de inicio tardío. La epidemiología exacta se desconoce, pero en la literatura internacional existen cerca de 300 casos reportados y se ha reportado una incidencia de 1/200,000 recién nacidos vivos en Francia2.
A continuación se presentan tres casos de niños con SHACC tratados en la Clínica de Trastornos Respiratorios del Dormir del Instituto Nacional de Enfermedades Respiratorias (INER) de México.
2Casos clínicos2.1Caso 1Paciente masculino de 12 años de edad, con de peso 20.6kg y talla 125cm, que fue hospitalizado por neumonía adquirida en la comunidad, y posteriormente enviado a este servicio por hipercapnia crónica y cor pulmonale. No manifestaba síntomas de disautonomías; su sueño fue descrito como normal, sin ronquido ni apneas presenciadas. Se documentó por oximetría (SpO2) insuficiencia respiratoria crónica grave del 57%, dióxido de carbono exhalado (EtCO2) de 47mmHg (Tabla 1), gasometría con pH 7.4, PaCO2 45.4mmHg, PaO2 44.1mmHg, HCO3 27.8mmol/l, SaO2 79.9%. En la polisomnografía (PSG) se encontró EtCO2 máximo de 82mmHg (Figura 1); las pruebas de función respiratoria y radiografía de tórax no evidenciaron alteraciones. En el electrocardiograma se encontró con sobrecarga del ventrículo derecho, ecocardiograma con crecimiento de cavidades derechas y presión sistólica de la arteria pulmonar de 34mmHg. El resto de los estudios de laboratorio no presentaron alteraciones. Se inició tratamiento con ventilación mecánica no invasiva a presión positiva (VMNI), y se mantuvo estable por 9 meses, pero suspendió la VMNI y falleció 6 meses después por insuficiencia respiratoria aguda y cor pulmonale. No se realizó análisis genético.
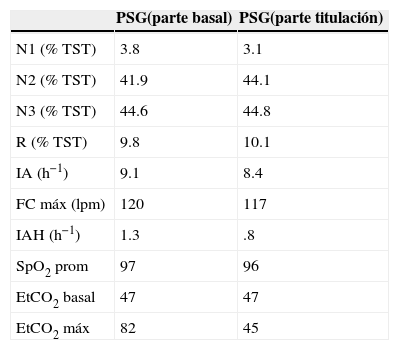
Polisomnografía del caso 1 en formato de noche dividida
| PSG(parte basal) | PSG(parte titulación) | |
|---|---|---|
| N1 (% TST) | 3.8 | 3.1 |
| N2 (% TST) | 41.9 | 44.1 |
| N3 (% TST) | 44.6 | 44.8 |
| R (% TST) | 9.8 | 10.1 |
| IA (h−1) | 9.1 | 8.4 |
| FC máx (lpm) | 120 | 117 |
| IAH (h−1) | 1.3 | .8 |
| SpO2 prom | 97 | 96 |
| EtCO2 basal | 47 | 47 |
| EtCO2 máx | 82 | 45 |
Comparación entre la parte basal y la de titulación. Destaca la hipercapnia grave en la parte basal resuelta en la fase de titulación.
PSG: polisomnografía; N: sueño de no movimientos oculares rápidos; R: sueño de movimientos oculares rápidos; TST: tiempo total de sueño; IA: índice de alertamientos; FC: frecuencia cardiaca (en latidos por minuto); IAH: índice de apnea hipopnea; SpO2: oximetría; EtCO2: dióxido de carbono exhalado.
Nota: En la parte basal se colocó oxígeno por puntas nasales a un flujo de 1 l/min lo que normalizó la SpO2; la parte de titulación se llevó a cabo sin oxígeno suplementario.
 que demuestra hipercapnia grave durante el sueño (EtCO2: dióxido de carbono exhalado).")
Paciente masculino de 1 año y 3 meses, producto de un embarazo a término y normoevolutivo, con crecimiento y desarrollo adecuados para su edad, que ingresó a hospitalización por neumonía adquirida en la comunidad e insuficiencia respiratoria hipercápnica aguda grave, al parecer desproporcionada para su padecimiento. Presentó crisis convulsivas y respuesta exagerada a benzodiacepinas que requirió manejo con ventilación mecánica invasiva, y posterior dificultad para su retiro por hipercapnia. Durante su estancia en la unidad de cuidados intensivos se identificaron episodios nocturnos de taquicardia/bradicardia, taquipnea/bradipnea y diaforesis. La oximetría diurna del 91%, EtCO2 diurno de 44mmHg, EtCO2 nocturno de 66mmHg, PaCO2 diurna 41mmHg, PaCO2 nocturna 120mmHg. La resonancia magnética nuclear de cráneo, electroencefalograma y radiografía de tórax normales, y la ecocardiografía con ligero crecimiento de cavidades derechas. Se realizó PSG y se documentó hipoventilación nocturna grave. Se tituló un dispositivo de presión positiva (Tabla 2). Actualmente se encuentra estable y tratado con VMNI en modo binivel con frecuencia respiratoria de respaldo (ST). No se realizó análisis genético.
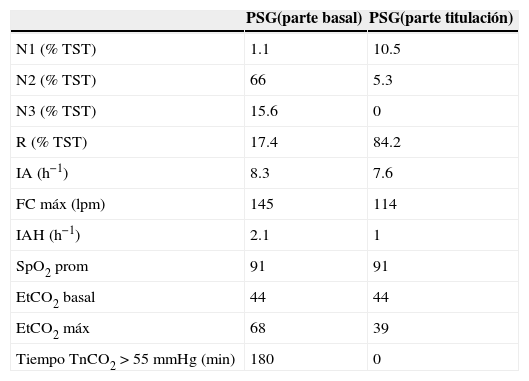
Polisomnografía del caso 2 en formato de noche dividida
| PSG(parte basal) | PSG(parte titulación) | |
|---|---|---|
| N1 (% TST) | 1.1 | 10.5 |
| N2 (% TST) | 66 | 5.3 |
| N3 (% TST) | 15.6 | 0 |
| R (% TST) | 17.4 | 84.2 |
| IA (h−1) | 8.3 | 7.6 |
| FC máx (lpm) | 145 | 114 |
| IAH (h−1) | 2.1 | 1 |
| SpO2 prom | 91 | 91 |
| EtCO2 basal | 44 | 44 |
| EtCO2 máx | 68 | 39 |
| Tiempo TnCO2>55mmHg (min) | 180 | 0 |
Comparación entre la parte basal y la de titulación. Destaca la hipercapnia grave en la parte basal resuelta en la fase de titulación.
PSG: polisomnografía; N: sueño de no movimientos oculares rápidos; R: sueño de movimientos oculares rápidos; TST: tiempo total de sueño; IA: índice de alertamientos; FC: frecuencia cardiaca (en latidos por minuto); IAH: índice de apnea hipopnea; SpO2: oximetría; EtCO2: dióxido de carbono exhalado; TcCO2: dióxido de carbono transcutáneo.
Nota: En la parte basal se colocó oxígeno por puntas nasales a un flujo de 1 l/min lo que normalizó la SpO2; la parte de titulación se llevó a cabo sin oxígeno suplementario.
Paciente masculino de 2 años y 8 meses de edad. Nació por cesárea tras 40 semanas de gestación; se calificó con Apgar 9/10. Al nacimiento estuvo internado durante 3 meses por hipoxemia atribuida a foramen oval permeable. Esta malformación se corrigió a los 6 meses de edad; sin embargo, persistió la hipoxemia. Durante el primer año de vida presentó tres episodios de insuficiencia respiratoria atribuidos a neumonías. Al año 9 meses de edad se documentó hipertensión arterial pulmonar durante el estudio por insuficiencia respiratoria hipercápnica. Durante una agudización de la insuficiencia respiratoria presentó crisis convulsivas, por lo que requirió ventilación mecánica invasiva por 9 días. En hospitalización se observaron apneas sin esfuerzo inspiratorio durante el sueño, desaturaciones e hipercapnia grave con PaCO2 de 110mmHg. En la valoración inicial se observó un niño con crecimiento y desarrollo adecuados para su edad, oximetría diurna en vigilia de 84% y EtCO2 de 64mmHg. En la PSG (Figura 2) se encontraron los siguientes hallazgos: arquitectura de sueño normal, índice de apnea hipopnea de 3.5 eventos por hora de sueño, CO2 transcutáneo (TcCO2) máximo de 160mmHg e hipoxemia corregida por suplemento de oxígeno a flujos bajos. Se instaló entonces tratamiento con un dispositivo de VMNI en modo binivel ST. Se solicitó estudio del gen PHOX2B el cual resultó positivo para el SHACC (genotipo 25/20). Después de 1 año de buena adherencia al tratamiento con un dispositivo de VMNI modo binivel ST, no ha requerido hospitalizaciones por ningún motivo. El intercambio de gases diurno es normal (SpO2 94%, EtCO2 34mmHg, gasometría con pH 7.4, PaO2 76mmHg, PaCO2 34mmHg, HCO3 24mmol/l, EB-2, SaO2 94%) y la ventilación nocturna está compensada. (Tabla 3).
, TcCO2 y SpO2. Destacan los valores de TcCO2 de 33mmHg (marco rojo) (SpO2: oximetría de pulso; TcCO2: dióxido de carbono transcutáneo).")
Poligrafía ventilatoria nocturna después de 1 año de seguimiento del caso 3. De arriba hacia abajo, las señales presentadas son flujo por neumotacógrafo, esfuerzo inspiratorio por banda en tórax y abdomen (pletismografía por inductancia), TcCO2 y SpO2. Destacan los valores de TcCO2 de 33mmHg (marco rojo) (SpO2: oximetría de pulso; TcCO2: dióxido de carbono transcutáneo).
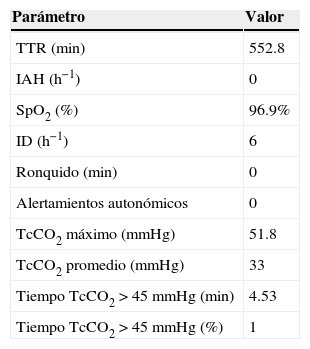
Poligrafía ventilatoria realizada al caso 3
| Parámetro | Valor |
|---|---|
| TTR (min) | 552.8 |
| IAH (h−1) | 0 |
| SpO2 (%) | 96.9% |
| ID (h−1) | 6 |
| Ronquido (min) | 0 |
| Alertamientos autonómicos | 0 |
| TcCO2 máximo (mmHg) | 51.8 |
| TcCO2 promedio (mmHg) | 33 |
| Tiempo TcCO2>45mmHg (min) | 4.53 |
| Tiempo TcCO2>45mmHg (%) | 1 |
Se midió el flujo por neumotacógrafo, esfuerzo por pletismografía por inductancia con banda en tórax y abdomen, TcCO2 y SpO2. Además del polígrafo se colocó un equipo de presión positiva binivel*. El estudio se realizó después de 1 año de tratamiento con ventilación mecánica no invasiva. Destacan los valores corregidos de la ventilación durante el sueño.
*Modo ST: con frecuencia respiratoria de respaldo, IPAP 13 cmH2O, EPAP 5 cmH2O, FR 21rpm, Timin 0.7 seg, Timax 1.2 seg, disparo y ciclado altos, TS 200 mseg, TD 100 mseg, FiO2 21%, a través de una mascarilla nasal infantil.
TTR: tiempo total de registro; IAH: índice de apnea hipopnea; SpO: pulsoximetría; ID: índice de desaturaciones; TcCO2: dióxido de carbono transcutáneo; IPAP: presión inspiratoria; EPAP: presión espiratoria; FR: frecuencia respiratoria; Timin: tiempo inspiratorio mínimo; Timax: tiempo inspiratorio máximo; TS: tiempo de subida; TD: tiempo de descenso; FiO2: fracción inspirada de oxígeno.
El SHACC es una enfermedad del sistema nervioso autónomo y del control de la respiración que se caracteriza por una disminución o ausencia de la respuesta ventilatoria a la hipercapnia o hipoxemia en ausencia de lesiones orgánicas específicas3–5. Fue descrita por primera vez en 1970 por Mellins y colaboradores6, y en 2003 se descubrió que las mutaciones del gen PHOX2B, localizado en el cromosoma 4p12, son responsables de este síndrome7,8.
En la literatura internacional se encuentran reportados cerca de 300 casos, y aunque no se conoce cuál es su incidencia en la población general, en 2005 Trang y colaboradores, a partir de la creación del registro de estos casos en Francia, estimaron una cifra de 1/200,000 recién nacidos vivos2. En los últimos años se ha incrementado el número de casos reportados, sobre todo en población caucásica. Sin embargo, este incremento se debe posiblemente a una mayor búsqueda y mejor identificación de los casos. Con mayor razón son necesarios estudios con grandes bases poblacionales y en diferentes grupos étnicos, para determinar la verdadera prevalencia de esta enfermedad5.
El mecanismo fisiopatológico exacto de la alteración en el control de la respiración no es claro aún. Se ha observado que la mayoría de los pacientes con esta enfermedad poseen un adecuado control de la ventilación durante la vigilia, e hipoventilación alveolar secundaria a la reducción del volumen corriente durante el sueño que es más severa durante la etapa de no movimientos oculares rápidos. En casos graves se ha observado hipoventilación tanto en vigilia como en sueño9.
En varios estudios se ha observado la ausencia en la respuesta de los quimiorreceptores centrales tanto a la hipercapnia como a la hipoxemia; incluso durante la vigilia y la respuesta de los quimiorreceptores periféricos parece estar menos afectada o intacta en niños capaces de tener una ventilación adecuada durante el estado de despierto. Estas observaciones apoyan la hipótesis de que existe una alteración en la integración de la información recibida por los quimiorreceptores en los centros de control respiratorio3,10-13.
El origen genético de la enfermedad se postuló con base en la concordancia de la enfermedad en gemelos homocigotos14,15. En 2003 se descubrió que la mutación del gen PHOX2B es causante de la enfermedad y tiene un patrón de herencia autosómico dominante, aunque en la mayoría de los casos se produce por mutación espontánea (90-95%)5,16. El gen PHOX2B codifica un factor de transcripción que regula la migración celular durante la formación de la cresta neural y el desarrollo del sistema nervioso autónomo (neurocristopatías)7,8,16. Esto explica la asociación que se ha observado entre las formas severas del SHACC y disautonomías, la enfermedad de Hirschsprung y tumores de la cresta neural16,17.
El 90% de los casos de SHACC es heterocigoto a la mutación de expansión repetida de polialaninas (PARM por sus siglas en inglés) en el gen PHOX2B, con un rango de 4 a 13 repeticiones adicionales (genotipos de 24/20 a 33/20). El número de mutaciones se asocia con la severidad de la alteración del control ventilatorio18,19. El 10% restante tiene otro tipo de mutaciones no relacionadas a PARM (NPARM), que se asocian con formas clínicas más graves de tumores de la cresta neural20.
Al momento del diagnóstico el cuadro clínico se puede dividir de acuerdo con la edad de inicio:
- •
Cuadro de inicio neonatal. La edad promedio de diagnóstico es de 3.5 meses2. Los pacientes con SHACC suelen presentar episodios intermitentes de cianosis o apnea al nacer y muchos requieren de ventilación mecánica inmediatamente después del parto, pero no suelen tener evidencia de alguna lesión neurológica que explique el trastorno ventilatorio. En otro grupo se presenta en los primeros meses de vida con episodios que amenazan la vida, e incluso paro respiratorio durante el sueño1,5.
En muchos pacientes durante el dormir se observa un patrón respiratorio regular pero superficial, con reducción del movimiento de la pared torácica y episodios de apneas centrales; además, pueden documentarse hipercapnia e hipoxemia que no se asocian con el incremento del esfuerzo ni de la frecuencia respiratoria. Tampoco se ven alertamientos y el paciente suele verse tranquilo. En muchos niños la hipoventilación se puede demostrar en estado despierto, pero con el crecimiento eventualmente respiran adecuadamente durante la vigilia por la maduración del sistema nervioso central, el crecimiento del aparato respiratorio (aumento de unidades alveolares y mejora de la relación volumen del espacio muerto/volumen corriente) y la disminución del tiempo total de sueño1,3,5. Estos cambios pueden dar una falsa y transitoria sensación de curación; sin embargo, la disminución/ausencia de respuesta ventilatoria a la hipercapnia/hipoxemia persiste, por lo que se sigue requiriendo el tratamiento.
En caso de no haber sido diagnosticados adecuadamente, estos niños desarrollan signos de insuficiencia cardiaca derecha e hipertensión arterial pulmonar. La hipoventilación suele empeorar, incluso con infecciones respiratorias leves, pues son incapaces de aumentar su respuesta ventilatoria a demandas incrementadas y no suelen presentar síntomas ni signos evidentes de insuficiencia respiratoria. Estos pacientes, en general, tienen mala respuesta a situaciones de ejercicio o estrés21.
Algunos infantes con SHACC pueden presentar signos de daño en órganos blanco, como cor pulmonale, convulsiones y retraso en el desarrollo psicomotor secundarios a hipercapnia e hipoxemia crónicas no detectadas previamente1,3,5. Además, se han descrito problemas de crecimiento y dismorfias craneofaciales; también se han encontrado niños con disautonomías, como diaforesis, taquicardia/bradicardia, taquipnea/bradipnea, respuesta pupilar alterada, alteración en la regulación de la temperatura, entre otras (Tabla 4)22–24.
Tabla 4.Cuadro clínico y manifestaciones del SHACC
Síntomas, signos y condiciones patológicas de alteración autonómica Respiratoria • Percepción de disnea ausente• Hipoventilación alveolar Cardiovascular • Perfusión periférica alterada• Variabilidad cardiaca disminuida• Pausas sinusales prolongadas• Síncope vaso-vagal Gastrointestinal • Dismotilidad esofágica• Constipación• Enfermedad de Hirschsprung Neurológica • Ansiedad disminuida• Percepción del dolor disminuida• Tumores de la cresta neural Ocular • Reflejos pupilares a la luz disminuidos• Anisocoria• Falta de mirada convergente• Estrabismo Otros • Sudoración profusa • Desregulación de la temperatura
- •
Cuadro de inicio tardío. La presentación en niños mayores o en la edad adulta es más rara aún. La gravedad de la enfermedad depende del genotipo, que usualmente se asocia con mutaciones cortas (23/20–25/20). Se suele observar en pacientes con algún tipo de alteración neurocognitiva, epilepsia, apnea del sueño o historia de hipoventilación durante la infancia. Puede detectarse ocasionalmente por hipoventilación luego de anestesia general o en infecciones respiratorias (Tabla 4)25–29.
Los criterios diagnósticos propuestos por la Academia Americana de Medicina del Sueño fueron actualizados en 2014 (Tabla 5)30,31.
Criterios diagnósticos del síndrome de hipoventilación alveolar central congénito
| Criterios Diagnósticos de SHACC |
| Deben cumplirse los criterios A y B |
| A. Presencia de hipoventilación relacionada al sueño |
| B. Mutación del gen PHOX2B |
| Notas: |
| 1. La presencia de hipoventilación relacionada al sueño puede asociarse con hipoventilación diurna o PaCO2 normal. |
| 2. La PSG demuestra hipercapnia severa y desaturación de oxígeno. Pueden observarse algunas apneas centrales pero el patrón predominante es la reducción en el flujo/volumen corriente. |
| 3. Aunque la condición se denomina congénita, algunos pacientes con genotipo PHOX2B pueden presentarse fenotípicamente más tarde, aun en la edad adulta, especialmente en presencia de un estresor como anestesia general o infección respiratoria severa. |
De acuerdo con la Clasificación Internacional de Trastornos de Sueño31.
SHACC: síndrome de hipoventilación alveolar central congénito; PaCO2: presión arterial de dióxido de carbono; PSG: polisomnografía.
- •
Se debe establecer la sospecha diagnóstica en aquellos pacientes con cuadro clínico sugestivo: hipoventilación sin causa orgánica evidente1,4,5, examen físico normal, alteraciones del patrón respiratorio e hipoventilación durante el sueño y signos de hipoxemia e hipercapnia crónicas.
- •
Detección de la mutación del gen PHOX2B. Debe realizarse en todo paciente con sospecha clínica. El 90% de los casos de SHACC son positivos para la mutación de este gen; sin embargo, ante una prueba negativa, un claro fenotipo clínico es suficiente para el diagnóstico4,5,32.
- •
Polisomnografía (PSG). Con capnometría a través de medición transcutánea o exhalada continua, pues es necesario estudiar el nivel de hipoventilación durante el sueño9,33,34.
- •
Evaluaciones adicionales. Deben realizarse exámenes adicionales para evaluar la severidad y cronicidad de la enfermedad, además de identificar las alteraciones autonómicas asociadas1,4:
Eritrocitosis: hematocrito y hemoglobina sanguíneos.
Acidosis respiratoria crónica: gases arteriales, bicarbonato sérico.
Hipertensión pulmonar–cor pulmonale: ecocardiograma, electrocardiograma, péptido natriurético cerebral (BNP) en sangre.
Enfermedad de Hirschsprung: enema baritado y biopsia de mucosa rectal en pacientes con constipación o distensión abdominal.
Arritmias cardiacas: monitoreo Holter.
Tumores de la cresta neural: imágenes de tórax y abdomen ante la sospecha de tumor neural.
Retardo en el desarrollo o aprendizaje: evaluación neurocognitiva completa.
El objetivo principal en el tratamiento del SHACC es asegurar la adecuada oxigenación y ventilación durante el sueño y la vigilia para mejorar el pronóstico a largo plazo al reducir el riesgo de desarrollar cor pulmonale, hipertensión arterial pulmonar y daño neurológico secundarios a la hipoxia crónica1,4,5.
El manejo de los defectos del desarrollo del tubo neural y las disautonomías es complejo y requiere de la participación de equipos multidisciplinarios.
3.3Soporte ventilatorioDado que en los niños con SHACC la respuesta ventilatoria a la hipoxemia e hipercapnia no mejora con el crecimiento ni con el uso de fármacos estimulantes del centro respiratorio, se requiere del uso de algún sistema de soporte ventilatorio35. Estos dispositivos deben ajustarse para obtener niveles de PaCO2 entre 30 y 40mmHg y saturación de oxígeno mayor al 95%5. Existen varios tipos de dispositivos de ventilación. Usualmente se utiliza la ventilación a presión positiva por traqueostomía y la VMNI a través de mascarilla. La titulación de los parámetros ventilatorios se debe realizar mediante PSG en la cual se puede comprobar la corrección del intercambio gaseoso durante el sueño.
- •
Ventilación a presión positiva por traqueostomía (VPPT). Se emplea frecuentemente en infantes y niños pequeños. Se suele utilizar en estas edades hasta que se logre la maduración del aparato respiratorio y del sistema nervioso central. Se considera el manejo más seguro para garantizar la adecuada ventilación y oxigenación. Es necesario un equipo portátil con una batería interna. Se requiere de al menos dos cuidadores entrenados en el uso del equipo y en el cuidado de la traqueostomía. Se recomienda el uso de tubos de traqueostomía sin balón para permitir una fuga suficiente para el uso de una válvula de Passy–Muir® que estimule el desarrollo del habla y evite estenosis subglótica. De igual forma, el equipo debe ser capaz de compensar la fuga y, a la vez, garantizar una adecuada insuflación pulmonar5,36,37.
- •
Ventilación no invasiva con presión positiva binivel. Se realiza a través de mascarillas nasales u oronasales. Es menos costosa y fácil de utilizar que la VPPT; sin embargo, no es aconsejable en pacientes que requieran 24h de ventilación o presiones altas. El uso prolongado de mascarillas para VMNI puede producir lesiones en piel e hipoplasia facial en pacientes muy jóvenes. Debido a que los pacientes con SHACC no pueden gatillar adecuadamente al ventilador, el modo ventilatorio utilizado debe ser capaz de dar una frecuencia respiratoria de respaldo. También es útil para reemplazar a la VPPT en pacientes con curso clínico estable que sean capaces de cooperar con el uso de VMNI —usualmente a partir de la edad escolar— y en los que se planee el retiro de la cánula de traqueostomía. En estos casos, las presiones requeridas para ventilación suelen ser mayores a las utilizadas en VPPT debido a que la VMNI debe superar la resistencia de la vía aérea superior38–41.
- •
Otros sistemas de soporte ventilatorio. Se ha descrito el uso de sistemas de ventilación a presión negativa por medio de chalecos o caparazones de tórax42, así como la colocación de marcapasos diafragmáticos. Estos últimos suelen asociarse con la VMNI, y ambos dispositivos pueden alternarse durante el día y la noche, respectivamente43–45.
El uso del soporte ventilatorio requiere seguimiento médico periódico y ajustes en los parámetros ventilatorios mediante PSG al menos una vez al año, sobre todo en niños pequeños debido a su crecimiento46. Los niños con SHACC tienen mayor riesgo de padecer alteraciones neurocognitivas que dificultan el aprendizaje47. En adolescentes debe vigilarse el consumo de alcohol y drogas, ya que estas sustancias pueden deprimir aún más el control de la respiración y aumentar el riesgo de muerte48,49. También el manejo inadecuado de la ventilación incrementa el riesgo de daño causado por la hipoventilación e hipoxemia crónicas50.
El único factor identificado asociado con mortalidad es el sexo: los varones presentan un mayor riesgo. En pacientes sin soporte ventilatorio el pronóstico es malo, especialmente con tasas de mortalidad elevadas en los primeros 3 meses de vida, lo que confirma que el centro respiratorio es más inestable a esta edad y que el soporte ventilatorio debe instaurarse tan pronto como se sospeche el diagnóstico51. La mortalidad global, según diferentes series, varía entre el 8 y 38%. Las principales causas de muerte son neumonía, cor pulmonale descompensado y aspiración; la mortalidad es más alta en pacientes sin soporte ventilatorio o con soporte inadecuado52,53.
El SHACC es un padecimiento genético raro pero cada vez más frecuentemente identificado en los servicios de neumopediatría y en las clínicas de sueño. Requiere un alto nivel de sospecha en pacientes (especialmente pediátricos) con insuficiencia respiratoria hipercápnica sin origen aparente. El diagnóstico temprano es importante para el inicio del soporte ventilatorio, lo que ayudará a prevenir el desarrollo de complicaciones y reducir la mortalidad.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
FinanciamientoNinguno.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.