













Introducción: La calcinosis tumoral es una enfermedad rara, cuya prevalencia es difícil de determinar debido a lo aislado de los casos. Se caracteriza por depósitos periarticulares, únicos o múltiples, de cristales de calcio y fosfatos. Esto es debido a una falla en la inhibición de la recaptura de fosfatos a nivel de los túbulos proximales, lo que causa hiperfosfatemia. No está descrito un tratamiento óptimo y el reducido número de casos dificulta la evaluación de los resultados.
Caso clínico: Se presenta el caso de una paciente de sexo femenino de 10 años de edad, con un cuadro clínico caracterizado por aumento de volumen en ambos codos y en el dedo índice de la mano derecha. Se realizó diagnóstico de calcinosis tumoral hiperfosfatémica. Se dio tratamiento quirúrgico y médico, sin presentar recidiva en los primeros seis meses de seguimiento.
Conclusiones: La calcinosis tumoral es una entidad rara que debe sospecharse al tener un paciente con calcificaciones periarticulares únicas o múltiples. El tratamiento médico debe orientarse a restablecer el equilibrio calcio-fósforo. El tratamiento quirúrgico se utiliza por razones estéticas y funcionales.
Background: Tumoral calcinosis is a rare disease whose prevalence is hard to determine due to the scarcity of the reported cases. This disease is distinguished by single or multiple periarticular deposits of calcium and phosphate due to a failure in the phosphate recapture inhibition at the proximal renal tubules, resulting in hyperphosphatemia. There is no optimal treatment described because there are a reduced number of cases, which makes it difficult to assess the results.
Case report: We present the case of a 10-year-old female with a growing mass in both elbows and index finger of the right hand. Diagnosis was made of hyperphosphatemic tumoral calcinosis. Surgical and medical treatment were given, with no relapse during a 6-month follow-up.
Conclusions: Tumoral calcinosis is a rare entity that should be considered when a patient has a single or multiple periarticular calcifications. Medical treatment is oriented to restoring the calcium-phosphate balance. Surgical treatment should be offered for aesthetic and functional reasons.
1. Introducción
La calcinosis tumoral es una enfermedad rara. Fue descrita por primera vez con este nombre en 1943, por Inclán y colaboradores1, aunque hay reportes de casos similares reportados en 1889, por Duret2, y en 1953, por Teutschlaender quien la llamó lipocalcinogranulomatosis3. Su prevalencia es difícil de determinar debido a lo aislado de los casos. Se ha observado una mayor presentación en pacientes de raza negra, sin predominio de sexo, durante la primera y segunda décadas de la vida.
En condiciones normales, los niveles séricos de fosfato dependen de tres factores: la absorción intestinal, el almacenamiento en el esqueleto y la reabsorción renal. Estos procesos, a su vez, están regulado por tres factores endócrinos: el factor de crecimiento de fibroblastos 23 (FGF23), la hormona paratiroidea (PTH) y el calcitriol4 (1,25(OH)2D3).
La absorción de fosfato en el intestino está mediada por el 1,25-(OH)2D3. El fosfato sérico es captado por las células por medio de cotransportadores de sodio-fosfato de tipo II y III, para realizar funciones como la síntesis de ADN y lípidos de membrana, entre otras5. Otra parte del fosfato sérico es almacenado en el esqueleto en forma de cristales de hidroxiapatita. La reabsorción de fosfato en los riñones es mediada por la PTH y el FGF23, y se lleva a cabo en los túbulos proximales, por medio de los cotransportadores de sodio-fosfato tipo II y tipo III. El cotransportador de sodio-fosfato tipo II es responsable del 70-80% de la reabsorción de fosfato4,5.
El FGF23 es un factor endócrino con actividad hormonal que reduce la concentración de fosfato sérico, disminuyendo su absorción intestinal y la reabsorción en los túbulos proximales. El FGF23 se produce en los osteocitos, los osteoblastos y las células osteoprogenitoras, y es regulado por el PHEX (homólogo de la endopeptidasa reguladora de fosfato ligado al cromosoma X) y la DMP1 6 (proteína de matriz de la dentina 1). Antes de que FGF23 sea secretado a la circulación, pasa por una etapa de O-glucosilación mediada por la proteína GALNT3 (polipéptido N-acetilgalactosaminil trasferasa 3), para poder transformarse en su forma activa. En los túbulos proximales, FGF23 se une, junto con Klotho alfa (α-KL), al receptor FGFR1c (receptor del factor de crecimiento de fibroblastos 1c). La activación de este receptor disminuye la expresión de los cotransportadores de sodio-fosfato tipo IIa y IIc, inhibiendo la reabsorción de fosfato. El FGF23 también reduce la síntesis de 1,25(OH)2D3, disminuyendo la absorción de calcio y fosfato intestinal5.
La calcinosis tumoral familiar hiperfosfatémica es una enfermedad autosómica recesiva que se caracteriza por depósitos periarticulares de cristales de calcio y fosfato en tejidos blandos, debido a un aumento de la concentración de fosfato sérico7. La hiperfosfatemia se debe a que no se inhibe la recaptura de fosfato a nivel de los túbulos proximales por medio del FGF235. Se han descrito tres mecanismos diferentes para el mal funcionamiento de FGF23. En el tipo 1 existe una mutación homocigota en el gen que codifica la proteína GALNT3, por lo que no se lleva a cabo la glucosilación de FGF23. Los niveles séricos de FGF23 activo se encuentran disminuidos mientras que los niveles de FGF23 no glucosilado están elevados. En el tipo 2, una mutación homocigota en FGF23 resulta en un nivel sérico elevado de FGF23 que no es funcional. En el tipo 3 se presenta una mutación homocigota del gen que codifica α-KL, la enzima que, junto con FGF23, activa al receptor FGFR1c, por lo que, aunque haya FGF23 intacto circulante, no se puede transmitir su señal a través del receptor5. En conjunto, estos mecanismos permiten que haya un aumento en la reabsorción de fosfato renal y, como resultado, hiperfosfatemia. Entre los hallazgos clínicos que se pueden observar debido a la hiperfosfatemia se encuentran la formación de depósitos de calcio y fosfato periarticulares y en tejidos blandos, calcificaciones en la pulpa dental, calcificaciones en los párpados, calcificaciones vasculares, nefrocalcinosis, y densidad mineral ósea8. Las tumoraciones periarticulares, no dolorosas, aumentan de tamaño progresivamente (durante meses o años), generalmente en la caras extensoras de las articulaciones, sin causar limitación en el movimiento de las mismas. Las articulaciones más afectadas son la cadera, el hombro y los codos, aunque cualquier articulación puede verse afectada. La piel que se encuentra subyacente a las lesiones puede presentar tractos fistulosos, con drenaje de material caseoso de manera intermitente que cesa espontáneamente9.
Las imágenes radiográficas típicamente muestran calcificaciones periarticulares amorfas, quísticas, multilobuladas10.
Los exámenes de laboratorio no muestran alteraciones en los niveles de calcio sérico. Sin embargo el fósforo sérico puede encontrarse normal o aumentado, con absorción renal de fósforo aumentada. Los niveles de hormona paratiroidea se encuentran dentro de rangos normales. La función renal esta preservada.
Los hallazgos anatomopatológicos muestran masas cálcicas blanco amarillentas lobuladas. Al microscopio, se presentan calcificaciones rodeadas por reacción inflamatoria crónica de cuerpo extraño con células gigantes10.
Debido a su baja incidencia, no se han establecido guías de tratamiento. Sin embargo, se han reportado casos donde el tratamiento médico ha sido exitoso11-13. Por un lado, se busca disminuir la absorción intestinal de fosfato mediante la restricción de la dieta (a 400 mg al día más hidróxido de aluminio y/o sevelamer)4. Para inhibir la reabsorción de fosfato renal se recomienda el uso de acetazolamida4,5. Cuando las lesiones causan dolor, deformidad o limitación en el movimiento, se recomienda la resección quirúrgica de los depósitos14-18. Sin embargo, las lesiones tienden a recurrir si persiste la alteración metabólica4, por lo que la resección se recomienda en conjunto con el tratamiento médico19,20.
2. Caso clínico
Se presenta el caso de una paciente de sexo femenino de 10 años de edad, proveniente de Matamoros, Tamaulipas, México. Previamente sana, sin antecedentes personales de importancia, sin antecedentes de endogamia en la familia o familiares con lesiones similares a las que ella presenta. Acudió a consulta por presentar un aumento de volumen en la cara extensora del codo derecho desde hacía un año, aumento de volumen en la cara extensora de codo izquierdo desde hacía seis meses, salida de material caseoso de manera intermitente del codo izquierdo desde hacía tres meses, y aumento de volumen en el dedo índice de mano derecha dos meses antes. Durante este periodo de tiempo nunca presentó fiebre, pérdida de peso, debilidad, mialgias, dolor ni limitación a la movilización de las articulaciones afectadas. Recibió dos esquemas de antibióticos sin presentar mejoría.
Previamente, se obtuvo el consentimiento escrito de la madre de la paciente para el reporte de este caso y sus imágenes.
A la exploración física se encontraron ambos codos con un aumento de volumen en la cara extensora de los mismos, de consistencia blanda, sin limitación en los rangos de movimiento de flexión ni extensión, sin dolor a la movilización (figs. 1 y 2). En el codo izquierdo se observaron múltiples lesiones, que corresponden con los tractos de salida de material caseoso, sin datos de infección y sin salida de material caseoso en ese momento (figs. 3 y 4). En el pulpejo del dedo índice de la mano derecha se observaron lesiones blanco amarillentas a través de la piel, de consistencia blanda, dolorosas a la palpación. En el resto de las articulaciones no se encontraron lesiones, tumoraciones ni dermatosis. Se buscaron, intencionadamente, datos de calcinosis en dientes y ojos, sin hallazgos significativos.
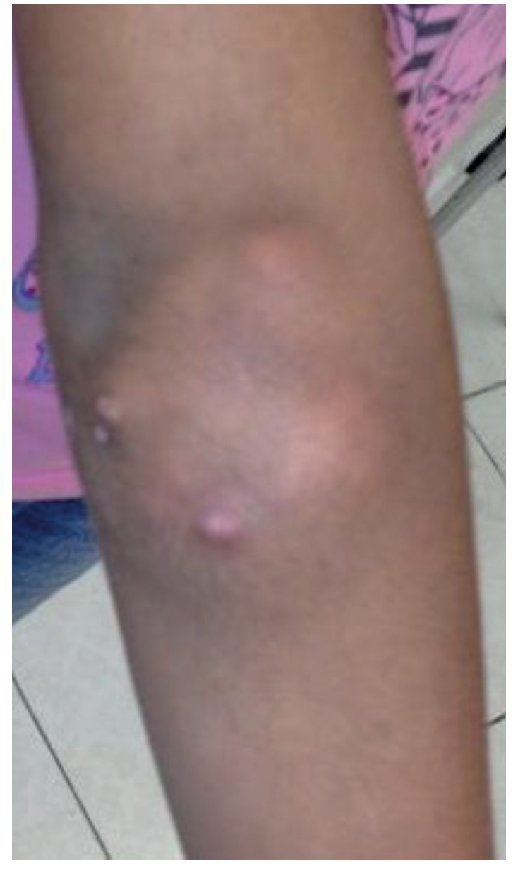
Figura 1. Vista frontal de codo derecho.
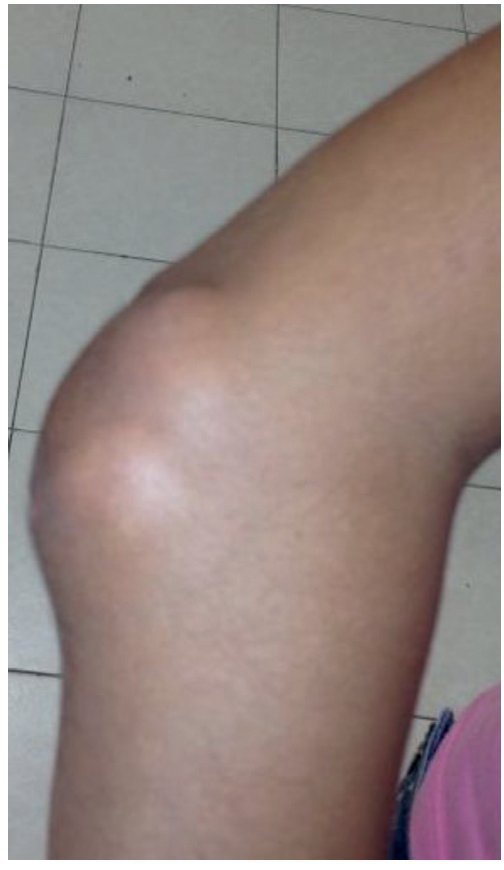
Figura 2. Vista lateral de codo derecho.
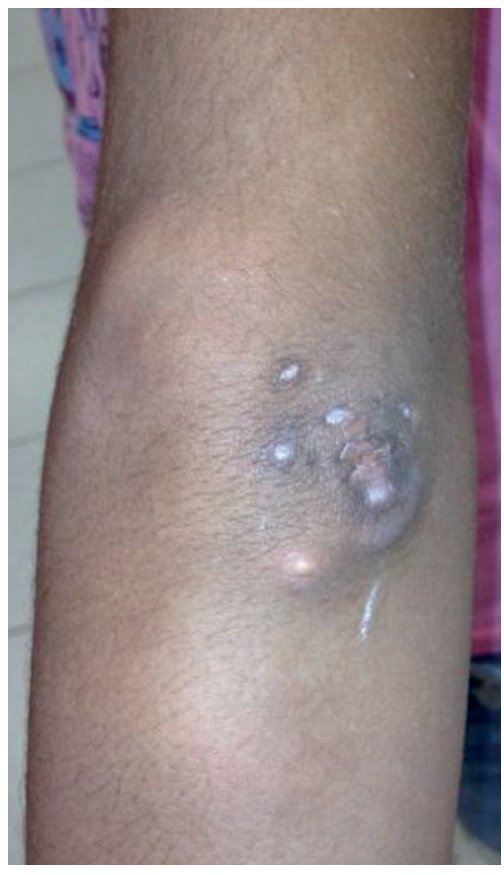
Figura 3. Vista frontal de codo izquierdo.
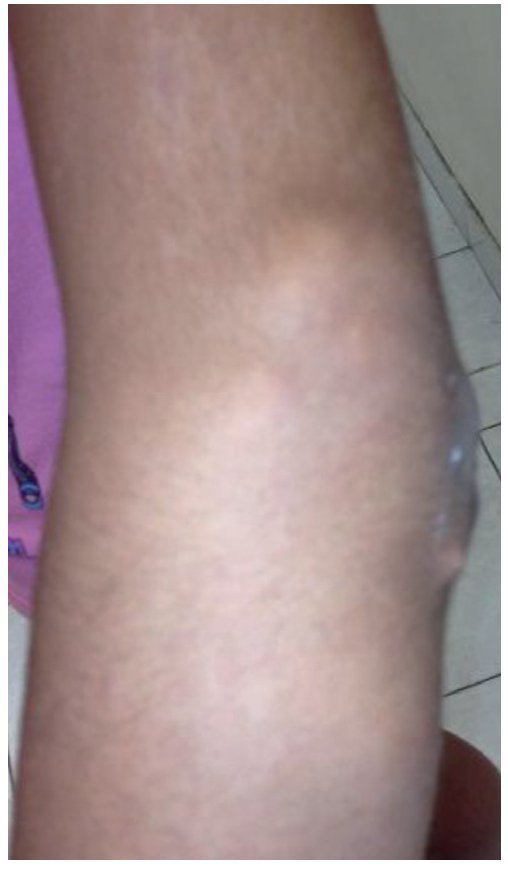
Figura 4. Vista lateral de codo izquierdo.
Se tomaron radiografías de los codos (figs. 5 y 6) y de la mano derecha (fig. 7) en las que se observaron conglomerados de múltiples opacidades homogéneas, redondeadas, separadas por líneas radiolúcidas, que no afectan ni dependen de las estructuras óseas adyacentes. Se solicitaron también radiografías de tórax y abdomen, ecocardiograma, ultrasonido renal y TAC de cráneo simple en busca de calcificaciones. Todos los estudios se reportaron sin hallazgos significativos.
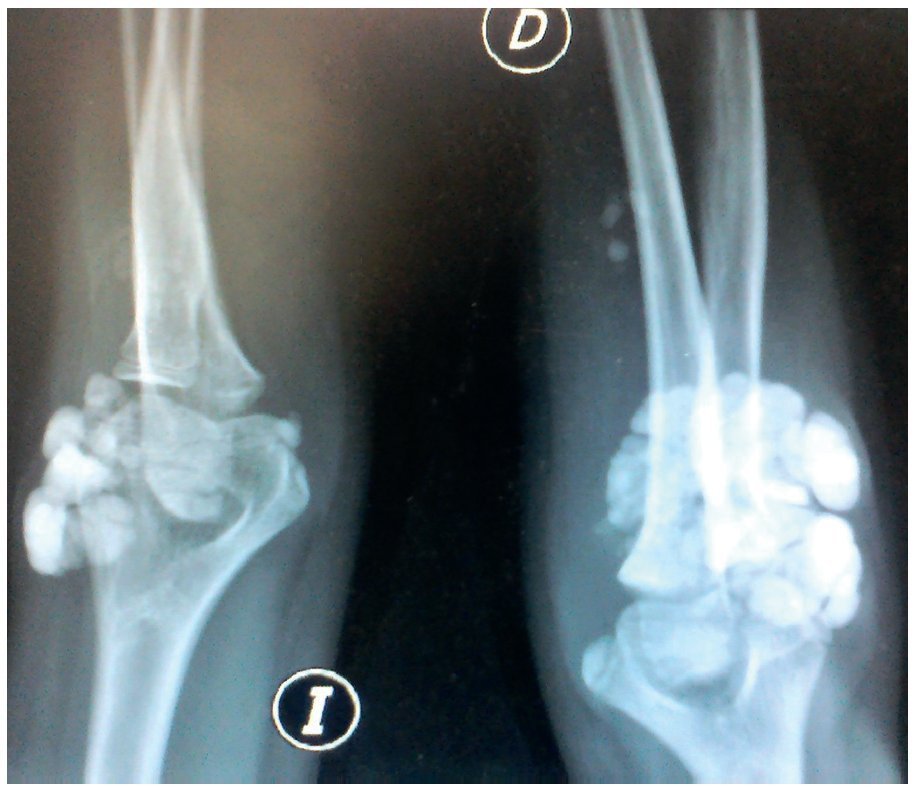
Figura 5. Radiografía anteroposterior de ambos codos (I: izquierdo; D: derecho).
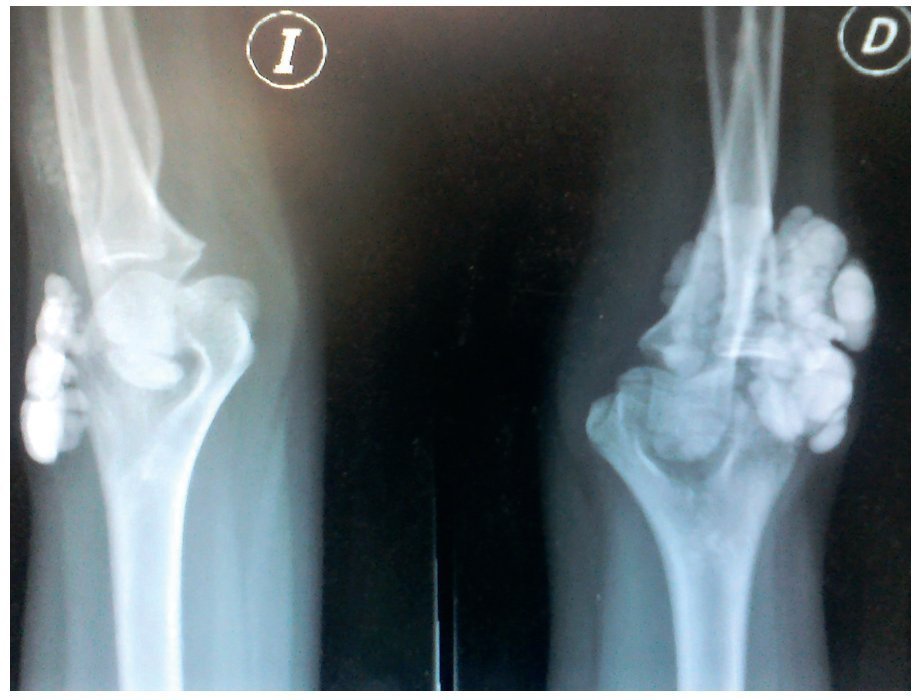
Figura 6. Radiografía lateral de ambos codos (I: izquierdo; D: derecho).
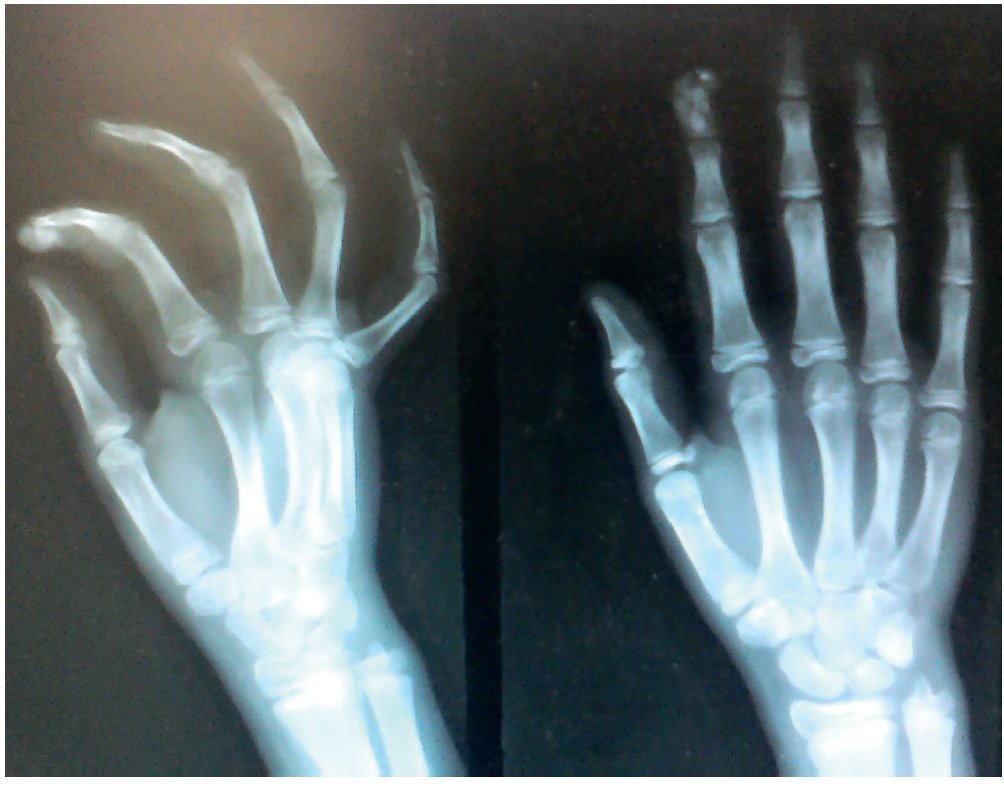
Figura 7. Radiografía anteroposterior y lateral de mano derecha.
Los exámenes de laboratorio resultaron con creatinina sérica normal (0.28 mg/dl), sodio sérico normal (139 mmol/l), potasio normal (4.1 mmol/l), normocalcemia (9.8 mg/dl), hiperfosfatemia (5.9 mg/dl), hormona paratiroidea intacta normal (27.5 pg/ml), fósforo en orina de 24 horas disminuido (307 mg/dl), umbral máximo de reabsorción tubular de fosfato de 5.72 mg/dl, reabsorción tubular de fósforo aumentada (97.1%), y con una tasa de filtración glomerular corregida por superficie corporal de 293.79 ml/min/m2. Se realizó el diagnóstico de calcinosis tumoral hiperfosfatémica.
Se realizó la resección quirúrgica completa de las calcificaciones de los codos, enviando las piezas para su examen anatomopatológico. A la exploración macroscópica se reportaron fragmentos de color gris-blanquecino, aspecto lobulado y consistencia laxa (fig. 8). La superficie de corte de la sección fue heterogénea, y se observaron múltiples nódulos de color blanquecino, aspecto caseoso y consistencia friable en un fondo fibroso (fig. 9). La exploración histopatológica con tinción de hematoxilina y eosina reportó grandes masas de calcificación depositadas en el estroma conjuntivo, rodeadas por reacción de cuerpo extraño y actividad fibroblástica (fig. 10). No se observaron osteocitos ni osteoblastos (figs. 11).
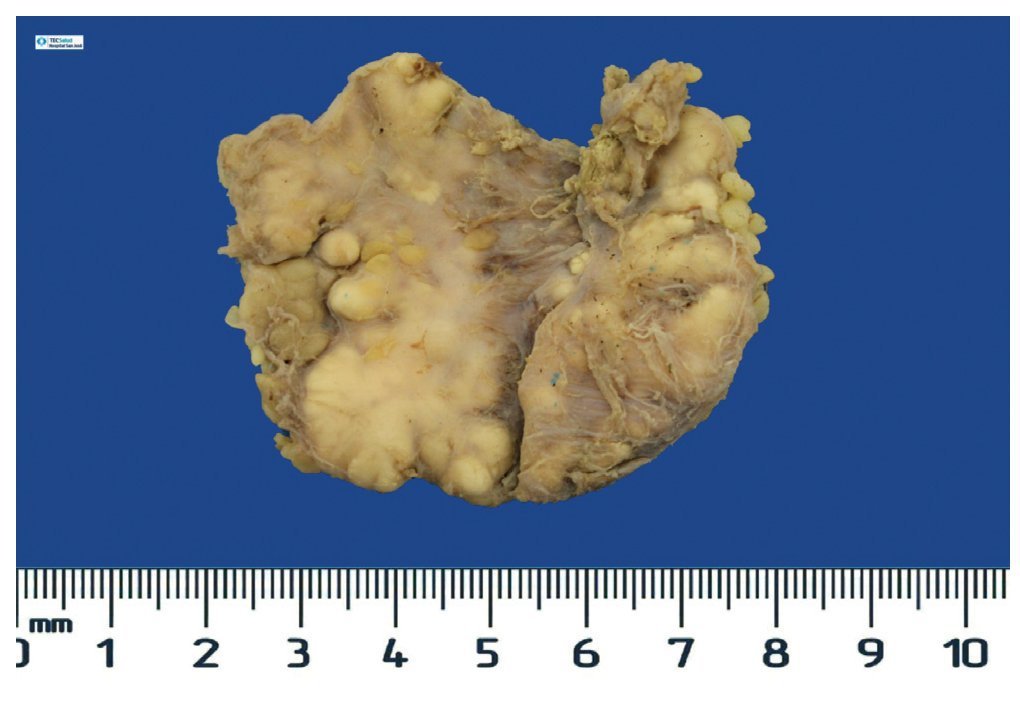
Figura 8. Tumoración irregular de tejidos blandos de codo derecho, de 24.2 g y 6.0 × 4.5 × 2.0 cm.
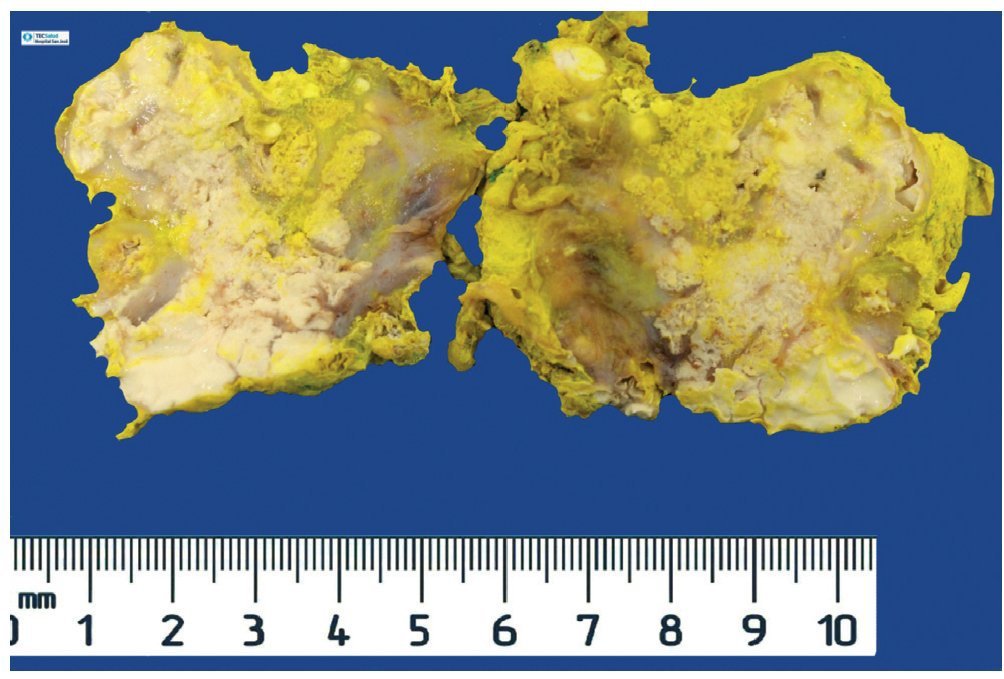
Figura 9. Al corte, la tumoración es heterogénea, de aspecto multilobulado y de consistencia friable.
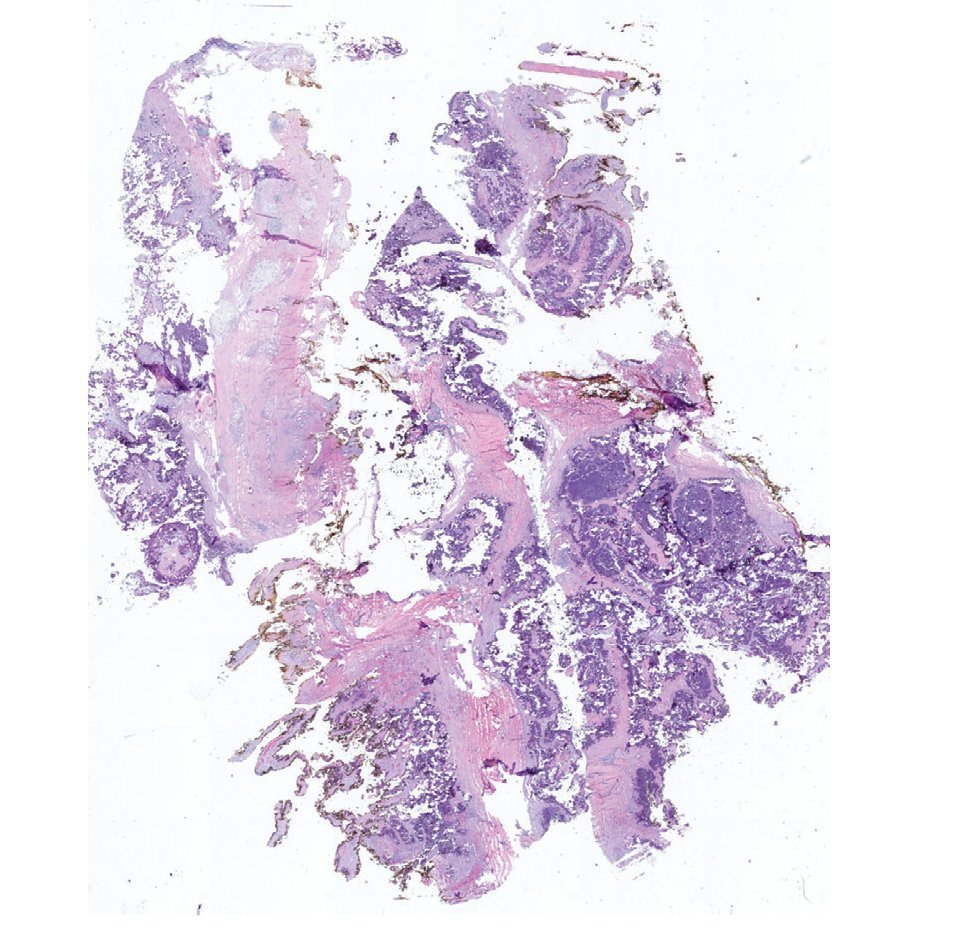
Figura 10. Corte representativo de la lesión que demuestra abundante calcificación distrófica rodeada por tejido fibroso. Borde de resección circunferencial en amarillo (1.25× HE).
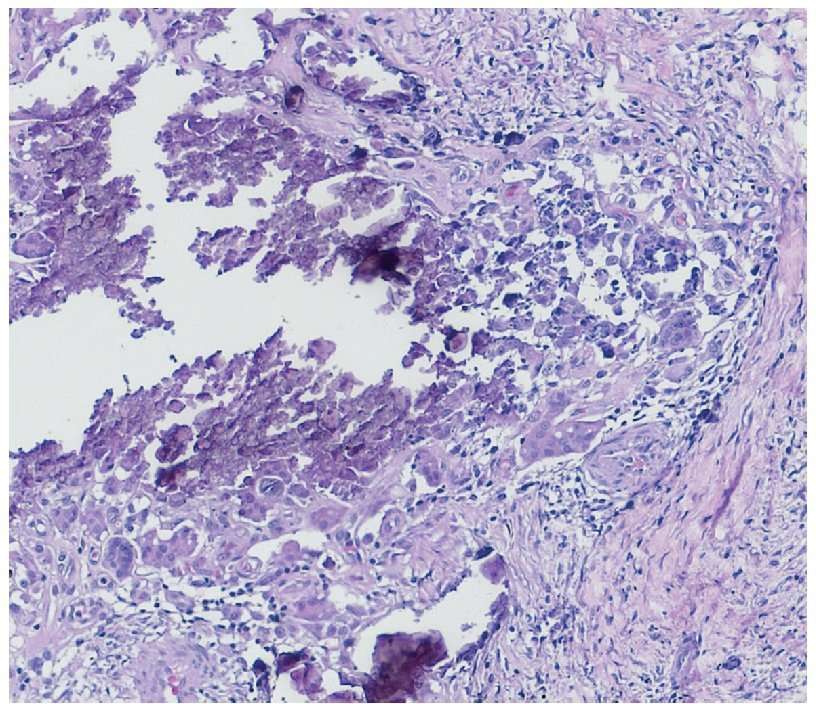
Figura 11. Calcificación distrófica rodeada por células gigantes multinucleadas de tipo cuerpo extraño e inflamación crónica (40× HE).
Posterior a la resección de las calcificaciones de los codos, se egresó con tratamiento con 10 ml de hidróxido de aluminio y magnesio cuatro veces al día, previo a los alimentos, y una dieta baja en fósforo. Se dio seguimiento telefónico seis meses después del inicio del tratamiento. Se reportó que no había recidiva de las calcificaciones de los codos, y que la calcificación que se encontraba en el dedo índice de la mano derecha había disminuido de manera importante, hasta ser casi imperceptible. Se continuó con el mismo manejo.
3. Discusión
La presencia de calcificaciones periarticulares, únicas o múltiples, es un hallazgo raro en pediatría. En el diagnóstico diferencial debe sospecharse la calcinosis tumoral junto con otras patologías que pueden compartir sus características radiológicas, distribución, tamaño y morfología, principalmente enfermedades de tejido conectivo (dermatomiositis, esclerodemia, lupus, síndrome de CREST), enfermedades neoplásicas (sarcoma sinovial, osteosarcoma) e insuficiencia renal crónica.
Sin embargo, la historia clínica de la calcinosis tumoral es muy particular —tumoraciones periarticulares que aumentan de tamaño progresivamente en meses o años, no dolorosas, generalmente en las caras extensoras de las articulaciones, que no causan limitación en el movimiento de las mismas—, al igual que sus hallazgos radiológicos (calcificaciones periarticulares, lobuladas, separadas por líneas radiolúcidas en las radiografías simples) y los resultados de laboratorio (normocalcemia e hiperfosfatemia o normofosfatemia, con funciones renal y paratiroidea normales).
Uno de los diagnósticos diferenciales de la calcinosis es la esclerodermia. La esclerodermia es una enfermedad autoinmune del tejido conectivo que involucra daño a la piel, a los vasos sanguíneos, a los músculos y a los órganos internos. En pacientes en edad pediátrica, las características principales son piel endurecida e inicio antes de los 16 años. Se divide en dos categorías: la escleroderma localizada, donde hay involucramiento de la piel pero no de órganos internos ni del sistema vascular; y la esclerodermia sistémica, la cual sí involucra órganos internos, particularmente esófago, tracto gastrointestinal, corazón, pulmones y riñones21.
Para el diagnóstico de esclerodermia sistémica se utilizan ciertos criterios preliminares21.
Entre los criterios mayores se encuentran los siguientes:
• Esclerosis o induración de tejidos
• Esclerodactilia
• Fenómeno de Raynaud
Entre los criterios menores se encuentran los siguientes:
• Cambios vasculares (cambios ungueales, capilares, úlceras digitales)
• Afectación gastrointestinal (disfagia, reflujo gastroesofágico)
• Afectación renal (crisis renal, hipertensión de nuevo inicio)
• Afectación cardiaca (arritmias, insuficiencia cardiaca)
• Afectación pulmonar (fibrosis pulmonar, hipertensión pulmonar)
• Afectación musculoesquelética (roce articular, artritis, miosisits, calcificaciones subcutáneas)
• Afectación neurológica (neuropatía, síndrome del túnel del carpo)
• Afectación serológica (anticuerpos antinucleares, anticuerpos específicos de esclerosis sistémica)
La paciente de este caso solamente presentó un criterio menor para esclerodermia sistémica, por lo que la probabilidad de este diagnóstico era baja.
En la esclerodermia localizada, las lesiones comúnmente están confinadas a la dermis, con poco involucramiento profundo. Se han publicado casos aislados donde se involucran órganos internos en pacientes con esclerodermia localizada; sin embargo, las lesiones dermatológicas predominan en estos casos21.
El diagnóstico se realiza con base en el cuadro clínico y la biopsia de la piel o tejidos subcutáneos donde se encuentre la lesión. Se pudo descartar este diagnóstico debido a que el cuadro clínico no siguió el patrón de la esclerodermia, específicamente por los signos dermatológicos que se requieren para su diagnóstico21.
Otro diagnóstico diferencial que se debe descartar es la dermatomiositis. La dermatomiositis juvenil es la miopatía inflamatoria idiopática juvenil más común. Es una enfermedad autoinmune caracterizada por una vasculopatía sistémica con miositis, debilidad muscular progresiva y hallazgos dermatológicos. No se sabe con certeza cuál es la etiología de esta enfermedad, pero se cree que es una combinación de predisposición genética y de factores ambientales22.
El diagnóstico se realiza por medio de los siguientes criterios:
• Involucramiento muscular: debilidad muscular progresiva y simétrica. Puede haber disfagia e involucramiento respiratorio.
• Biopsia de músculo: fibras de necrosis tipo I y II, fagocitosis, regeneración con basofilia, núcleos sarcolémicos, vesículas grandes, nucléolos prominentes, atrofia con distribución perifascicular, fibras de tamaños variables, exudados inflamatorios comúnmente perivasculares.
• de enzimas musculares: CPK, aldolasa, AST, deshidrogenasa láctica.
• Electromiograma: unidades motoras polifásicas cortas y pequeñas, fibrilaciones, ondas positivas agudas, descargas repetitivas de alta frecuencia.
• Cambios dermatológicos: rash en heliotropo con edema periorbitario, signo de Gottron (dermatitis eritematosa y escamosa en el dorso de las manos), involucramiento de las rodillas, codos, maléolo medial, cara, cuello y parte proximal del tórax.
• El diagnóstico es definitivo cuando se tienen 3 o 4 criterios, además de los hallazgos dermatológicos. Si se cuenta con dos criterios más hallazgos dermatológicos, se dice que el diagnóstico es probable22.
La calcinosis distrófica es una complicación característica de la dermatomiositis juvenil, reportada en 30-70% de los pacientes con el diagnóstico ya establecido. Las calcificaciones se encuentran en codos y rodillas, principalmente. Pueden causar dolor local, contractura de la articulación y úlceras en la piel suprayacente. Esta complicación se debe a que los músculos dañados secretan calcio a las vesículas de la matriz y promueven la mineralización con acumulación de hidroxiapatita22.
Aunque esta paciente presentó lesiones similares a las lesiones encontradas en la calcinosis distrófica, no cumplió los criterios para el diagnóstico de dermatomiositis juvenil.
También se debe descartar lupus eritematoso sistémico (LES), una enfermedad autoinmune del tejido conectivo que puede afectar cualquier parte del cuerpo. Lo más común es que los pacientes en edad pediátrica presenten síntomas sistémicos como fiebre, caída de pelo, fatiga, pérdida de peso, linfadenopatía y hepatoesplenomegalia23. Los órganos más afectados en niños son la piel (rash malar, fotosensibilidad, lesiones vasculares, eritema palmar y plantar, fenómeno de Raynaud y eritema anular), sistema musculoesquelético (artritis, artrialgias, tenosinovitis) y renal23.
Las lesiones articulares relacionadas con LES se caracterizan por un derrame articular de leve a moderado, con disminución en el rango de movimiento, y que aumenta de dolor por las mañanas23. Ya que la paciente no cumplió con este cuadro, se descartó esta patología.
Una vez realizado el diagnóstico de calcinosis tumoral, debe de buscarse el tratamiento ideal para el paciente. Debe tomarse en cuenta que, actualmente, la teoría más aceptada es que la enfermedad está causada por una disfunción en la excreción de fosfatos a nivel de los túbulos proximales renales, lo que causa hiperfosfatemia secundaria y calcificaciones masivas en regiones periarticulares. El tratamiento debe estar orientado a restablecer el metabolismo calcio-fósforo, y el tratamiento quirúrgico debe de utilizarse por razones estéticas y funcionales. Aún después de la disminución de las calcificaciones con tratamiento médico o la resección quirúrgica de las mismas, existe riesgo de recidiva, especialmente si no se realizó una resección completa y en pacientes con hiperfosfatemia. Por ello, es importante dar seguimiento a los pacientes en busca de recidiva de las calcificaciones existentes o aparición de nuevas. También es importante el seguimiento a estos pacientes, ya que su pronóstico cardiovascular es pobre debido a la calcificación vascular acelerada que sufren.
En el caso que se presentó en este trabajo, se decidió el tratamiento quirúrgico en los codos por razones estéticas, realizándose una resección completa de las calcificaciones, y se inició tratamiento médico con dieta baja en fósforo e hidróxido de aluminio y magnesio para disminuir la absorción intestinal de fosfatos. Con ello, disminuyó la calcificación del dedo índice de la mano derecha, por lo cual no se realizó la resección quirúrgica de esta calcificación.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
Recibido el 7 de noviembre de 2013;
aceptado el 2 de junio de 2014
* Autor para correspondencia.
Correo electrónico:dr.abegalindo@gmail.com (A. Galindo Gómez).