





El síndrome de muerte súbita asociada a una imagen electrocardiográfica similar al bloqueo de rama derecha (BRD) del haz de His y elevación no-isquémica del segmento ST fue descrito originalmente por médicos italianos de la Universidad de Padua cuyos hallazgos fueron publicados en 1988 en una revista francesa y en 1989 en una americana1–3. Esta entidad es ahora mundialmente conocida gracias a la descripción que hicieron un poco después los hermanos Pedro y Josep Brugada4, quienes no solo contribuyeron a llamar la atención sobre el problema, sino que, junto con su hermano menor Ramón, aportaron conocimientos acerca de sus mecanismos de producción. Como se discutió en los primeros artículos, el BRD no es una lesión del haz derecho sino un patrón electrocardiográfico similar, de clasificación incierta en los ochenta. La razón de por qué este síndrome recibió dicho nombre es multifactorial pero una explicación reciente5 señala: «That the Brugada brothers were the only authors of the original report, combined with the interesting phonetics of Brugada and the long title of this newly described entity, probably contributed to its quick adoption».
Hasta donde se ha podido establecer, el primer electrocardiograma (ECG) con dicho patrón morfológico fue descrito en 1953 por Osher y Wolff6, quienes informaron una anormalidad electrocardiográfica dinámica, que simulaba un infarto agudo del miocardio en un hombre saludable. Es interesante lo que ellos escribieron: «This is apparently due to prolongation of the depolarization process by right bundle block or possibly focal block with delayed activation of a portion of the right ventricle: unusually early onset of repolarization may also play a role».
El síndrome, tal como fue descrito originalmente por los médicos italianos, es decir, la presencia de un evento clínico (muerte súbita) asociado a un patrón ECG característico, y no solo la presencia de dicho patrón en un trazo aislado, fue descrito inicialmente como la asociación entre ese patrón electrocardiográfico peculiar en las primeras derivaciones precordiales, un evento cardiaco importante y una sutil enfermedad cardiaca orgánica del ventrículo derecho (VD). Se interpretó que dicho patrón ECG se originaba por un retardo de la conducción a nivel infrahisiano y del tracto de salida del VD (TSVD), según se demuestra por los registros de superficie, los endocárdicos y las señales promediadas1. Se consideró que la causa básica era la sustitución fibroadiposa2,3. Las anormalidades se caracterizan por lesiones dentro del sistema principal de conducción (haz de His y rama derecha) e incluyen atrofia, fibrosis y adiposis de la pared ventricular derecha, en especial del TSVD7,8. En dichos informes iniciales también se identificaron otras características clínicas relevantes tales como: la historia familiar, el predominio en los hombres y la presencia de fibrilación ventricular (FV) fácilmente inducible mediante estimulación eléctrica del TSVD1–3.
Después del artículo de Josep y Pedro Brugada4, quienes no encontraron anormalidad cardiaca subyacente con el uso del ecocardiograma convencional, la mayor parte de la literatura (más de 3,000 artículos hasta el presente) se enfocó a la investigación de una base funcional. El principal dato sugerente de un desorden funcional fue la observación de cambios dinámicos del ECG, presente en más de la mitad de estos sujetos, más tarde hubo algunos experimentos de laboratorio y hallazgos genéticos9,10. Recientemente se han publicado evidencias de un defecto de conducción y una anormalidad orgánica subyacente en el síndrome de Brugada (SBr) y Nademanee discutió las teorías orgánicas y funcionales y demostró que las anormalidades en las derivaciones precordiales se deben a un trastorno de la conducción en el TSVD, el cual se observó fibrótico a nivel epi- y endocárdico, e incluso puede eliminarse mediante ablación con catéter11.
Cerca de 3 décadas después de su primera descripción por los médicos italianos, se han retomado las teorías iniciales, es decir, se vuelve a establecer que existe una anormalidad estructural subyacente en la entidad. Todo síndrome es una entidad clínica y por ello se necesitan datos clínicos para una correcta identificación antes de llevar a cabo experimentos de laboratorio y de realizar una estratificación de riesgo3,9. Esta visión clínica no siempre ha sido observada por los médicos y muchos jóvenes, por lo demás saludables, han sido investigados de modo invasivo (estudio electrofisiológico mediante cateterismo) e incluso han sido tratados con implante de desfibriladores tras un estudio electrofisiológico positivo.
Este trabajo intenta analizar las teorías orgánicas subyacentes en el síndrome y la historia de su descubrimiento, con la discusión de los datos publicados. La colección completa de los artículos sobre el síndrome (de uno de los autores, BM), excluye algunos artículos chinos y analiza todos los casos publicados en quienes se ha asociado una anormalidad estructural al síndrome, hasta enero del 2016. No se trata de una revisión sistemática sobre todos los aspectos del síndrome, con atención limitada a su anormalidad funcional, cuestiones ampliamente descritas en revisiones anteriores3. Se presentan evidencias que confirman los hallazgos iniciales del grupo de Padua para ofrecer al lector una nueva perspectiva del SBr en relación con el trasfondo anatómico.
Pistas para identificar una enfermedad cardiaca estructural latente en pacientes afectados por un verdadero síndromeLa historia médica olvidada. Los estudios anecdóticos de Gotoh y Kirshner et al.12,13 evidenciaron lesiones histológicas significativas del sistema de conducción en la gran mayoría de los asiáticos que morían súbitamente en la noche (en quienes el síndrome se sospechó retrospectivamente, sin ECG recuperado). Es interesante que muchas de estas víctimas tuvieron fiebre (se conoce que ella incrementa el patrón ECG del síndrome) en forma previa a la muerte súbita12. En años recientes no se ha tomado en cuenta o fue desatendida la experiencia de patólogos anteriores. Después de esos artículos, no existen estudios de autopsia detallados de las víctimas de muerte súbita en el Lejano Oriente, donde la enfermedad es relevante. Es posible que los patólogos no quieran impugnar el estado oficial (sin datos basados en la evidencia), en cuanto a que todas las víctimas de muerte súbita tuvieron corazones normales8. Si se examina la literatura médica, no se encuentra un estudio necrópsico detallado de un corazón normal en un paciente con el síndrome, ni en el Lejano Oriente ni en los países occidentales.
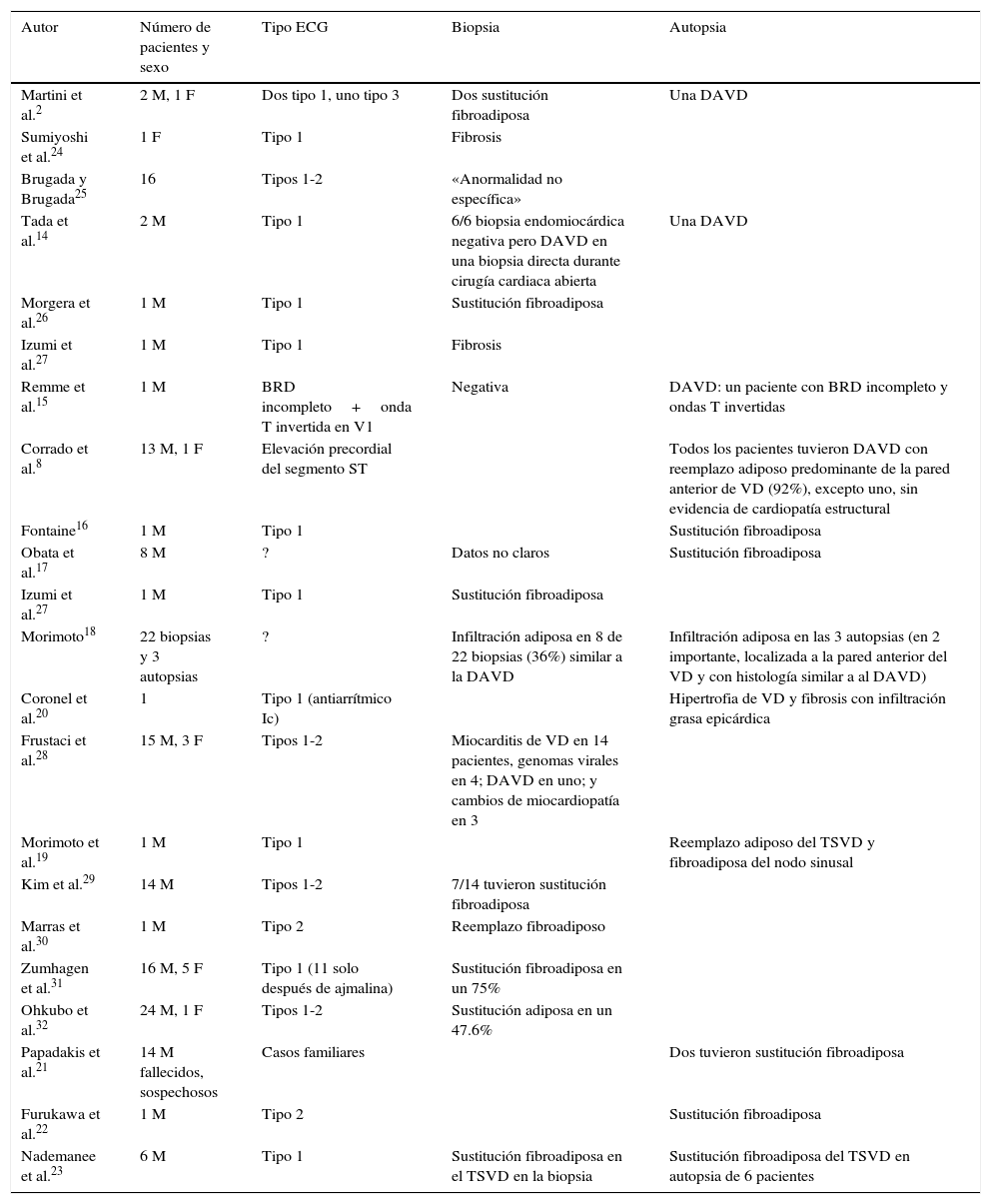
Datos basados en la evidencia en estudios necrópsicos detalladosEl estudio necrópsico es el estándar de oro para hacer un diagnóstico correcto y para cuantificar la especificidad y la sensibilidad de otras investigaciones, en tanto la biopsia parece menos importante por la imposibilidad o la peligrosidad de examinar solo las áreas patológicas localizadas del VD, del TSVD y del sistema de conducción. Se insiste en que el síndrome es una de las mayores causas de muerte súbita, a pesar de lo cual en la actualidad hay muy pocas autopsias en estos pacientes (tabla 1) y todas han evidenciado una sustitución fibroadiposa en especial en el TSVD y en el sistema de conducción.
Pacientes con el síndrome y alguna anormalidad en la biopsia o en la necropsia
| Autor | Número de pacientes y sexo | Tipo ECG | Biopsia | Autopsia |
|---|---|---|---|---|
| Martini et al.2 | 2 M, 1 F | Dos tipo 1, uno tipo 3 | Dos sustitución fibroadiposa | Una DAVD |
| Sumiyoshi et al.24 | 1 F | Tipo 1 | Fibrosis | |
| Brugada y Brugada25 | 16 | Tipos 1-2 | «Anormalidad no específica» | |
| Tada et al.14 | 2 M | Tipo 1 | 6/6 biopsia endomiocárdica negativa pero DAVD en una biopsia directa durante cirugía cardiaca abierta | Una DAVD |
| Morgera et al.26 | 1 M | Tipo 1 | Sustitución fibroadiposa | |
| Izumi et al.27 | 1 M | Tipo 1 | Fibrosis | |
| Remme et al.15 | 1 M | BRD incompleto+onda T invertida en V1 | Negativa | DAVD: un paciente con BRD incompleto y ondas T invertidas |
| Corrado et al.8 | 13 M, 1 F | Elevación precordial del segmento ST | Todos los pacientes tuvieron DAVD con reemplazo adiposo predominante de la pared anterior de VD (92%), excepto uno, sin evidencia de cardiopatía estructural | |
| Fontaine16 | 1 M | Tipo 1 | Sustitución fibroadiposa | |
| Obata et al.17 | 8 M | ? | Datos no claros | Sustitución fibroadiposa |
| Izumi et al.27 | 1 M | Tipo 1 | Sustitución fibroadiposa | |
| Morimoto18 | 22 biopsias y 3 autopsias | ? | Infiltración adiposa en 8 de 22 biopsias (36%) similar a la DAVD | Infiltración adiposa en las 3 autopsias (en 2 importante, localizada a la pared anterior del VD y con histología similar a al DAVD) |
| Coronel et al.20 | 1 | Tipo 1 (antiarrítmico Ic) | Hipertrofia de VD y fibrosis con infiltración grasa epicárdica | |
| Frustaci et al.28 | 15 M, 3 F | Tipos 1-2 | Miocarditis de VD en 14 pacientes, genomas virales en 4; DAVD en uno; y cambios de miocardiopatía en 3 | |
| Morimoto et al.19 | 1 M | Tipo 1 | Reemplazo adiposo del TSVD y fibroadiposa del nodo sinusal | |
| Kim et al.29 | 14 M | Tipos 1-2 | 7/14 tuvieron sustitución fibroadiposa | |
| Marras et al.30 | 1 M | Tipo 2 | Reemplazo fibroadiposo | |
| Zumhagen et al.31 | 16 M, 5 F | Tipo 1 (11 solo después de ajmalina) | Sustitución fibroadiposa en un 75% | |
| Ohkubo et al.32 | 24 M, 1 F | Tipos 1-2 | Sustitución adiposa en un 47.6% | |
| Papadakis et al.21 | 14 M fallecidos, sospechosos | Casos familiares | Dos tuvieron sustitución fibroadiposa | |
| Furukawa et al.22 | 1 M | Tipo 2 | Sustitución fibroadiposa | |
| Nademanee et al.23 | 6 M | Tipo 1 | Sustitución fibroadiposa en el TSVD en la biopsia | Sustitución fibroadiposa del TSVD en autopsia de 6 pacientes |
BRD: bloqueo de rama derecha; DAVD: displasia arritmogénica de ventrículo derecho; F: femenino; M: masculino; TSVD: tracto de salida de ventrículo derecho; VD: ventrículo derecho.
Primero, el grupo de Padua informó que un paciente con el síndrome (y una alteración del gen SCN5A) tuvo atrofia, fibrosis y adiposis de la pared libre del VD2,7,8, además de una lesión inusual del sistema de conducción, caracterizada por interrupción esclerótica de la rama derecha y por severa fibrosis del haz de His. Las lesiones de la pared libre, en particular a nivel infundibular, fueron compatibles con un segundo trastorno de conducción al final de la despolarización ventricular. En nuestra opinión, el retardo de la rama derecha puede explicarse por lesiones tanto del haz de His como de la rama derecha mientras que la elevación del segmento ST, por despolarización retardada del TSVD. No era fácil aceptar que el ST elevado en este síndrome correspondía a una anormalidad de la despolarización del TSVD pero la presencia de potenciales tardíos (PT), el empastamiento tardío del QRS y la onda épsilon ocasional, pudieran atribuirse a un defecto de conducción. No afirmamos que el VD enfermo del síndrome constituya una miocardiopatía arritmogénica del VD (displasia arritmogénica del VD [DAVD]) pues muchos de los pacientes no tienen los otros datos clínicos típicos ni el perfil genético, pero como se verá más adelante, existen ciertos datos compartidos entre ambas entidades no solo a nivel histopatológico, sino también a nivel genético.
Después de la detallada descripción del grupo de Padua, algunos otros estudios necrópsicos bien conducidos han confirmado una enfermedad ventricular derecha básica en el síndrome. Tada et al.14 describieron un paciente con sustitución adiposa del VD (no se estudió el sistema de conducción). Remme et al.15 informaron un caso con diagnóstico de DAVD en la investigación necrópsica, que se consideró clínicamente como un corazón normal (paro cardiaco por FV). Corrado et al.8 describieron evidencia necrópsica de DAVD en 13 de 14 jóvenes víctimas de muerte súbita en quienes el ECG evidenció ligera elevación del ST en las derivaciones precordiales. El reemplazo adiposo estuvo principalmente localizado en la pared anterior derecha. En 3 casos observaron fibrosis de la rama derecha.
En 1999 el doctor Pedro Brugada solicitó la opinión de Fontaine sobre el estudio anatomopatológico del corazón de una víctima del síndrome; él demostró con claridad que el paciente tenía datos como los que se encuentran en la DAVD. Este interesante caso fue presentado a la comunidad científica en un capítulo de libro16. En el 2000, Obata et al.17 publicaron 8 casos con el síndrome y sustitución fibroadiposa del VD (ECG no disponibles). Morimoto et al.18 describieron 3 casos fallecidos con una marcada infiltración adiposa del VD (TSVD). Esta serie no se publicó completa. Años más tarde19, el mismo autor publicó fotos de un caso con marcada sustitución en el TSVD. Se esperaba el BRD pero no las lesiones en el nodo sinusal (descritas por Gotoh en 1976)12. En el 2005, Coronel et al.20 publicaron el estudio histológico de un paciente trasplantado, con un patrón ECG tipo i después de administrar procainamida y que luego presentó ondas épsilon. Describieron el corazón derecho hipertrofiado con sustitución fibroadiposa, sobre todo del TSVD, responsable de un retardo de conducción y que excluía anormalidades de repolarización. En el 2013, Papadakis et al.21 publicaron 14 casos familiares sospechosos del síndrome que murieron súbitamente, al menos 2 de ellos tuvieron sustitución fibroadiposa miocárdica. En el 2014, Furukawa et al.22 encontraron en un paciente muerto súbitamente, con ECG tipo 1, una sustitución fibroadiposa del VD asociada a inflamación. Recientemente, Nademanee et al.23 informaron sobre 6 pacientes que murieron súbitamente (con historia familiar del síndrome) y 6 con ECG tipo 1 remitidos a ablación epicárdica durante una toracotomía y encontraron una relevante sustitución fibrótica del VD, en particular del TSVD. La necropsia carece de datos en cuanto al sistema de conducción. El artículo demuestra que la responsable de la elevación del ST es una conducción retardada en el TSVD a nivel epicárdico y no una enfermedad de canales iónicos («canalopatía»). Resulta interesante que en esta serie fueron raras las anormalidades del gen SCN5A.
Los datos basados en la evidencia de biopsia endomiocárdica del ventrículo derechoLa mayor parte de los estudios de biopsia que aparecen en la tabla 1 informan datos patológicos similares, incluso con resultados discutibles2,24–32. Las principales lesiones estructurales en la necropsia se observaron en el TSVD pero puede ser difícil y peligroso obtener especímenes adecuados de esta estructura. Los casos positivos quizás representen una enfermedad más difusa mientras que en los estudios negativos no puede descartarse una anormalidad localizada. En nuestra opinión, la biopsia no parece relevante para la detección de una enfermedad cardiaca estructural, en tanto otras investigaciones de superficie, los PT, el mapeo electroanatómico y el estudio de imagen por resonancia magnética pueden dar datos más seguros, si bien existen diferentes opiniones sobre su significado33, rediscutidas recientemente34.
El significado de las anormalidades estructurales del VD requiere más estudios. Las anomalías histológicas son similares a las de la DAVD y el TSVD es uno de los componentes del «triángulo de la displasia» descrito por Fontaine35; en general en la verdadera displasia las anormalidades estructurales predominan en el TSVD e interesan también el sistema de conducción infrahisiano. Aunque en el síndrome no se ha demostrado el papel de la inflamación, como en la DAVD, esta no puede excluirse. El desarrollo anormal del TSVD puede contribuir a la enfermedad36,37; esta última «hipótesis de Buenos Aires» es una de las 3 existentes sobre el síndrome y puede explicar no solo el síndrome sino otras arritmias ventriculares idiopáticas originadas en esta estructura36.
Pistas clínicas que indican una enfermedad cardiaca orgánicaLa historia clínica individual rara vez es útil para detectar enfermedad estructural en estos casos, pero sí lo es para detectar familiares afectados o para investigar la presencia de arritmias auriculares asociadas al síndrome. Es importante señalar que con frecuencia pueden verse patrones electrocardiográficos similares en familiares con eventos clínicos aunque también en los parientes de sujetos saludables y asintomáticos. Los datos de necropsia de un familiar fallecido pueden ser de utilidad21,23. La muerte súbita nocturna en las poblaciones del Lejano Oriente tendría alguna relevancia aunque no se ha probado que todas estas muertes se deban a SBr, por la casi total ausencia de ECG previos de los fallecidos. Por lo general la mayor parte de los pacientes con el verdadero síndrome no tienen síntomas relevantes antes del evento letal. El síncope durante el ejercicio es anecdótico3. Son frecuentes el predominio en los hombres (85%) y los síntomas mayores (muerte súbita o paro cardiaco reanimado) a edades de 30-40 años. Es rara una historia de palpitaciones y por lo general no hay disnea ni dolor torácico. Muy pocos pacientes tienen fiebre en las horas precedentes12.
Datos del electrocardiograma de superficie, del vectocardiograma y del electrocardiograma de señales promediadas que indican una enfermedad cardiaca orgánicaEl ECG es una herramienta muy sensible para detectar enfermedades cardiacas estructurales aunque su especificidad es discutible. En la actualidad, el único síndrome hereditario de muerte súbita que no tiene una enfermedad cardiaca estructural demostrable es probablemente el síndrome de QT largo, aunque no existen muchos estudios necrópsicos detallados en esta entidad. La mayor parte de las enfermedades cardiacas clásicas muestran signos eléctricos de hipertrofia y/o trastornos de conducción, que pueden clasificarse como diferentes grados de BRD o izquierda; desde el estudio pionero de Fontaine et al.38 existe evidencia de que la prolongación del complejo QRS y su fragmentación terminal pueden atribuirse a un trastorno de conducción localizado a diferentes áreas de las ramas. El segmento ST siempre se ha considerado parte del proceso de repolarización pero el grupo de Padua postuló que en este síndrome la elevación del ST puede ser un patrón peculiar de despolarización del TSVD1. En esta entidad la elevación en precordiales no es una anormalidad de repolarización como se ve en el infarto miocárdico o en la verdadera repolarización precoz, sino la expresión en el ECG de superficie de un trastorno de la conducción localizado e inusual. La mayor parte de la literatura reciente está de acuerdo con esto y estudios experimentales y en vivo demuestran que la elevación del ST en el ECG es provocada por un sustrato anormal y un trastorno de la conducción. Los datos preliminares del grupo de Amsterdam al respecto son promisorios39.
Aún existe mucha confusión entre la comunidad médica debido a que en el 2002 un grupo de expertos introdujo una clasificación que dividió el patrón ECG del síndrome en 3 tipos40. En primer lugar, se basaron solo en las 3 primeras derivaciones precordiales, olvidándose de las 9 restantes. Después de una década de artículos donde se hablaba de esta clasificación, muy poco se recordó que la misma es resultado del informe de un caso con alteraciones dinámicas de las derivaciones electrocardiográficas precordiales en un paciente resucitado3. Debió analizarse la prevalencia de este signo en una serie relevante de verdaderos pacientes, antes de proponer una clasificación que resultó desfavorable en algunos casos, debido a que fue empleada en exceso en individuos sanos y asintomáticos que presentaban alguno de estos 3 patrones encontrados de manera incidental (ya fuese de forma espontánea o en relación con fármacos), y que han sido sometidos a estudios invasivos e incluso a terapias inapropiadas. En fecha más reciente, expertos en ECG bajo la dirección de Bayés de Luna han propuesto una nueva clasificación41. Si se toman en cuenta las limitaciones de los consensos previos que describen el patrón de Brugada, en especial en relación con las diferencias entre los tipos 2 y 3, este grupo de expertos presentó un nuevo informe de consenso en el que establecen con mayor precisión nuevos criterios electrocardiográficos del síndrome. Como primicia, en los nuevos criterios solo se consideran 2 patrones, tipo 1 (clásico, de patrón cóncavo) y 2 (que une los patrones 2 y 3 del consenso anterior). En este importante documento se describen las características más importantes de estos 2 patrones y los puntos clave del diagnóstico diferencial con diversas condiciones que simulan un patrón de Brugada, tales como el BRD, el pectus excavatum, la DAVD y otros. Los autores describen criterios mucho más precisos que emplean ángulos, índices y análisis de la onda T que pueden ser consultados en dicha publicación.
El ECG del síndrome verdadero debe distinguirse de la simple presencia de un ECG similar en individuos saludables o en diferentes condiciones patológicas42, para evitar que se sobreestime la enfermedad y se realice un tratamiento inadecuado.
El ECG de la gran mayoría de los pacientes afectados por el verdadero síndrome se caracteriza por:
- 1.
Patrones morfológicos de los complejos QRS que semejan un BRD1,2, acompañados de una elevación característica del segmento ST (patrón en forma de domo) en las derivaciones precordiales V1 o V2 (figs. 1 y 2). A diferencia del verdadero BRD, la onda S se encuentra disminuida o ausente en V6, dando lugar a una mayor duración del complejo QRS en las derivaciones precordiales, con una falta de coincidencia de esta entre V1 y V6. Un intervalo PR prolongado, un intervalo HV largo y la desviación del eje eléctrico hacia la izquierda son indicadores de un trastorno de la conducción subyacente y son más frecuentes en aquellos con mutaciones en el gen SCN5A10,43. El ECG típico (tipo 1) es aquel con una elevación del ST en V1 con un patrón en forma de domo, ocasionalmente con una pequeña onda en forma de muesca aguda en el punto J (la unión del complejo QRS con el segmento ST). Este patrón en el ECG es erróneamente llamado onda J, diferente de la verdadera onda J descrita por Osborn y no debería llamársele así para evitar mayores confusiones44.
 Figura 1.
Figura 1.Derivaciones DI y V1 del primer paciente diagnosticado con el síndrome de muerte súbita, asociada a una morfología electrocardiográfica similar al bloqueo de rama derecha y elevación del segmento ST. Fue tomado poco después de un paro cardiaco reanimado mediante desfibrilación el 20 de noviembre de 1984. Es interesante que el cardiólogo lo interpretó como «taquicardia sinusal a 120lpm y bloqueo focal de rama derecha».
 ECG (1988) del primer paciente con el síndrome donde se observa un patrón característico (tipo 1) con un intervalo PR prolongado, desviación del eje a la izquierda, sin onda S en V6 (lo que traduce la ausencia de un verdadero bloqueo de rama derecha). B) ECG del mismo paciente (2005), donde se aprecian las ondas épsilon descritas inicialmente por Fontaine en la displasia arritmogénica de ventrículo derecho. No se observa onda S en V6. C) Acercamiento de las derivaciones V1 y V2 para mostrar más claramente las ondas épsilon de Fontaine.") Figura 2.
Figura 2.A) ECG (1988) del primer paciente con el síndrome donde se observa un patrón característico (tipo 1) con un intervalo PR prolongado, desviación del eje a la izquierda, sin onda S en V6 (lo que traduce la ausencia de un verdadero bloqueo de rama derecha). B) ECG del mismo paciente (2005), donde se aprecian las ondas épsilon descritas inicialmente por Fontaine en la displasia arritmogénica de ventrículo derecho. No se observa onda S en V6. C) Acercamiento de las derivaciones V1 y V2 para mostrar más claramente las ondas épsilon de Fontaine.
- 2.
El segmento ST más típico tiene una morfología en domo que es seguida por ondas T invertidas de ramas asimétricas en V1-V2, rara vez en V3. En pocas ocasiones se puede documentar un incremento de la elevación del segmento ST que preceda al evento arrítmico ventricular ni alteraciones espontáneas del ECG que puedan predecir la recurrencia de FV. En casos sin el patrón tipo 1, la inducción de la morfología tipo 1 con un fármaco se considera diagnóstica del síndrome. Aun cuando esta presunción es discutible, es la primera razón para colocar un desfibrilador automático implantable en sujetos asintomáticos45. En nuestra opinión esta indicación debe ser revisada.
En algunos casos43 el punto J puede estar fragmentado, tanto de forma espontánea como después de la administración de ajmalina3, con una forma que semeja mucho a las ondas épsilon descritas por Guy Fontaine38 (fig. 2). Se puede documentar una fragmentación de la porción terminal del complejo QRS (f-QRS) hasta en el 43%46–48 de presuntos pacientes, con mayor frecuencia en aquellos con patrón tipo 1, intervalos HV prolongados y FV.
- 3.
La elevación del segmento ST también puede mostrar una configuración de silla de montar (en el llamado tipo 2). En el verdadero síndrome, estos patrones pueden cambiar espontáneamente al tipo 1 por lo que se deben tomar varios ECG para ver si este patrón tipo 2 es constante o en algún momento pasa a tipo 1. La aparición de un patrón tipo 1 puede ser obtenida mediante la colocación de las derivaciones precordiales en los espacios intercostales-paraesternales superiores al cuarto espacio intercostal (donde habitualmente se registran las derivaciones V1 y V2), con el empleo de fármacos antiarrítmicos de clase 1C (p. ej., flecainida, ajmalina) e incluso muchos otros fármacos (ver sitio web www.brugadadrugs.org del Dr. Pieter Postema, de Holanda). Sin embargo, no debemos preocupar excesivamente a los individuos asintomáticos con un patrón tipo 2 debido a que ahora sabemos que incluso un verdadero síndrome, con patrón tipo 1, tiene muy poca probabilidad de presentar un evento arrítmico si el individuo es asintomático. En algunos pacientes se ha descrito la presencia de elevación del ST en derivaciones inferiores y laterales lo que se ha asociado con la presencia de eventos cardiacos mayores, en especial cuando también existe fragmentación terminal del QRS. En los pacientes con el síndrome, la presencia de onda J en múltiples derivaciones y la morfología horizontal del segmento ST después de la J puede indicar un sustrato altamente arritmogénico49,50.
El reconocimiento de que los bloqueadores de los canales de sodio empeoran transitoriamente la conducción y las alteraciones de la repolarización del SBr llevó al empleo de estos fármacos para desenmascarar el patrón ECG diagnóstico45. Sin embargo, el uso de fármacos antiarrítmicos de clase 1C para obtener el patrón ECG tipo 1 se ha popularizado incluso para personas con anormalidades precordiales inespecíficas de la porción terminal del QRS o del segmento ST. La comunidad médica abrazó esta nueva prueba diagnóstica, a pesar de que su especificidad es limitada45. Incluso en el momento actual, la mayoría de los estudios clínicos se realizan a personas asintomáticas aunque obtener el patrón tipo 1 con un fármaco no implica necesariamente que el sujeto tenga el síndrome, salvo que ello se pruebe por otras vías. En Europa, el 70% de los pacientes con «SBr asintomático» se diagnosticó después de una prueba positiva con ajmalina45. La razón más común para colocar un desfibrilador automático implantable en esos pacientes es «una prueba de ajmalina positiva más FV inducible durante el estudio electrofisiológico.»45. En nuestra opinión esta indicación debe ser revisada.
Un patrón tipo 1 pudo ser obtenido mediante fármacos en individuos asintomáticos con patrones 2 o 3 hasta en un 50-85% de una serie de individuos europeos por lo demás sanos3. Es interesante notar que hay más pruebas positivas en sujetos asintomáticos que en los casos sintomáticos o en los familiares de individuos con SBr, lo que plantea dudas científicas, éticas y legales. No hay evidencia de una correlación de la positividad de la prueba farmacológica con las mutaciones causantes de la enfermedad3. El efecto de estos medicamentos no es específico para el síndrome debido a que pacientes con miocardiopatía ventricular derecha e incluso sujetos normales pueden mostrar cambios similares en el ECG51. Una explicación más simple puede ser que la flecainida origine trastornos de conducción y no siempre en relación con la repolarización ni con los canales iónicos. Con su administración en sujetos sanos con una posible alteración latente de la conducción cabe esperar mayor retraso de la misma, sin que necesariamente esto implique el descubrimiento de un síndrome letal45,52, lo cual se demuestra mediante el registro de PT después de administrar dicho medicamento3.
La primera descripción de la utilidad de las derivaciones precordiales altas (por arriba del cuarto espacio intercostal) fue de Naccarella53 en 1993. Tras la publicación en 2001 de Sangwatanaroj et al.54 también se ha popularizado hacer un diagnóstico de SBr mediante el registro de V1 y V2 en espacios intercostales altos. Recientemente, Holst et al. han informado en 340 europeos saludables un 4.7% de prevalencia de tipo 2 y un 9.4% de tipo 3, con registros en los espacios intercostales superiores55. La obtención de un patrón tipo 1, diagnóstico de la enfermedad, es raro pero se puede hacer fácilmente con solo tomar las derivaciones precordiales a ambos lados del esternón en los espacios intercostales superiores56.
- 4.
El análisis vectocardiográfico se ha realizado rara vez desde su primera presentación1, pero muestra algunas características interesantes57: el final del bucle QRS no coincide con su principio, y su distancia depende del grado de elevación del ST. Hay retraso de la conducción posterior y superior derecha que probablemente indica un bloqueo parcial de las fibras que se acercan al TSVD (fig. 2, de Martini et al.3).
- 5.
Se pueden obtener PT en un ECG de señales promediadas hasta en un 70% de los casos3. Su presencia es menos frecuente en los patrones tipos 2 y 3. Su ausencia en casos con patrón tipo 1 indica que dicho patrón no se debe a una alteración de la conducción y es probable que exista alguna anomalía funcional subyacente (fig. 3). Los PT también pueden ser registrados a nivel del TSVD, durante el registro endocavitario, en el mapeo de superficie corporal y en registros epicárdicos directos. Se encuentran PT positivos con mayor frecuencia en pacientes que luego desarrollan eventos arrítmicos3,58,59.
; la ausencia de potenciales tardíos implica que dicho hallazgo podría ser secundario a un problema de la repolarización.") Figura 3.
Figura 3.Una estrategia simple para descartar un trastorno de la conducción como causa de la elevación del segmento ST. En este caso se trata de un hombre sano con elevación del segmento ST en precordiales derechas (que semeja un patrón tipo 1); la ausencia de potenciales tardíos implica que dicho hallazgo podría ser secundario a un problema de la repolarización.
- 6.
La fiebre puede inducir un patrón tipo 1 en el 2% de la población adulta60. En este estudio todos los individuos con patrón tipo 1 inducido por fiebre eran sujetos asintomáticos y se mantuvieron así durante 30 meses de seguimiento. La anomalía ECG es más fácil de ser inducida si se registran las precordiales derechas altas. Algunas series sugieren que la fiebre no se limita a originar cambios en el ECG, sino que en realidad puede provocar las arritmias fatales características del SBr61.
- 7.
Tanto el patrón ECG como los síntomas pueden aumentar con el predominio vagal (es decir, en la noche), pero los datos son insuficientes para confirmar esta hipótesis62. En individuos que no son atletas, los síntomas casi nunca se relacionan con el esfuerzo y la taquicardia ventricular (TV) durante la prueba de esfuerzo se ha reportado solo de forma anecdótica. Por lo que se refiere al patrón del ECG (tanto en los pacientes como en los sujetos asintomáticos), durante la prueba de esfuerzo el trazado suele normalizarse en el 25% del tipo 1, para luego volver a aparecer en la fase de recuperación. A pesar de estas evidencias limitadas, todas las guías y documentos de consenso desalientan a las personas con este ECG a practicar deportes3.
- 8.
Arritmias asociadas: los pacientes con el síndrome verdadero desarrollan FV o TV polimórfica1–3. Se observan extrasístoles ventriculares repetitivas monomorfas, en su mayoría con imagen de bloqueo de rama izquierda (BRI) (con eje superior o inferior), que inician las taquiarritmias ventriculares en más de 2 tercios de los casos3. La TV sostenida es una arritmia poco frecuente pero bien descrita43,63 en este síndrome, claramente asociada a un circuito estructural de reentrada. La mayoría de los casos muestran una morfología de BRI y un eje diferente. Los pacientes con arritmias ventriculares graves pueden tener pocas extrasístoles ventriculares durante el monitoreo Holter, cuya morfología es generalmente de BRI, sobre todo con un eje inferior3. Se pueden encontrar arritmias auriculares64 hasta en el 20% del verdadero síndrome, lo cual puede ser solo un marcador de enfermedad estructural más avanzada.

 ECG (1988) del primer paciente con el síndrome donde se observa un patrón característico (tipo 1) con un intervalo PR prolongado, desviación del eje a la izquierda, sin onda S en V6 (lo que traduce la ausencia de un verdadero bloqueo de rama derecha). B) ECG del mismo paciente (2005), donde se aprecian las ondas épsilon descritas inicialmente por Fontaine en la displasia arritmogénica de ventrículo derecho. No se observa onda S en V6. C) Acercamiento de las derivaciones V1 y V2 para mostrar más claramente las ondas épsilon de Fontaine.")
; la ausencia de potenciales tardíos implica que dicho hallazgo podría ser secundario a un problema de la repolarización.")
Un intervalo HV prolongado es casi la regla en pacientes con el ECG tipo 1, en especial si se asocia a la prolongación del intervalo PR y a la desviación del eje eléctrico2,4. Inicialmente se informaron potenciales fragmentados de QRS en el TSVD, coincidiendo con la elevación del segmento ST pero no recibieron mucha atención1. El intervalo HV prolongado es siempre un patrón de enfermedad cardiaca orgánica del sistema de conducción. La presencia de marcada fibrosis del sistema es la base de este hallazgo7,8,20. En otro paciente con enfermedad histológica cardiaca comprobada, los mapas de activación durante la estimulación de la zona del nodo auriculoventricular mostraron una propagación rápida normal de la activación del endocardio septal izquierdo y de todo el ventrículo izquierdo, mientras que en el TSVD se observó aglomeración de isocronos, indicativa de una conducción lenta. Toda la pared del VD (endocardio y epicardio) se activó relativamente tarde20.
El mapeo electroanatómico de alta resolución ha confirmado enfermedad cardiaca estructural derecha subyacente65, pero esta técnica por desgracia rara vez se utiliza durante el estudio electrofisiológico de rutina, que se ha dedicado principalmente a inducir arritmias ventriculares más que a entender su origen y su mecanismo básico3. Sin embargo, pocos pero relevantes estudios han indicado que este mapeo documenta en forma fiable áreas de conducción lenta, sobre todo a nivel del TSVD66. En esta zona se origina la mayor parte de las extrasístoles ventriculares, y es el mejor sitio para inducir FV con la estimulación eléctrica; su ablación por radiofrecuencia (endocardio y epicardio) puede ser «curativa», tanto experimental67 como clínicamente11,34. El TSVD es una estructura compleja desde el punto de vista embriológico36, donde se fusionan estructuras diferentes, creando una mezcla de propiedades estructurales y fisiológicas, distinta a otras regiones del miocardio. En estas áreas donde las peculiaridades de la despolarización y de la repolarización pueden estar presentes simultáneamente, se originan arritmias ventriculares benignas68 y malignas.
La inducción de la FV no es un criterio que permita el diagnóstico diferencial entre una anormalidad orgánica y una funcional. Por otro lado, su importancia es aún objeto de debate, en especial en relación con su papel en la estratificación del riesgo69,70.
Una pista importante que apoya una anormalidad estructural es la detección de espigas que indican la activación ventricular temprana dentro del sistema Purkinje derecho o del TSVD, como publicaron Haïssaguerre et al.71. La ablación de las extrasístoles ventriculares que usualmente se encuentran en ambas superficies endocárdica y epicárdica del TSVD, pueden abolir exitosamente las recurrencias11,34,72. Este es el hallazgo final que demuestra un sustrato subyacente y ha sido exitosamente realizado por los grupos de Brugada y Pappone.
Recientemente, el grupo de Amsterdam ha presentado una explicación fisiopatológica de la elevación del ST en el SBr39. Hasta el momento actual se han propuesto 3 hipótesis para explicar esta elevación: (1) un trastorno de la repolarización por un posible aumento de la dispersión transmural de la repolarización del VD o más específicamente del TSVD (estudios experimentales con registros monopolares endocárdicos y epicárdicos); (2) un trastorno de la despolarización, hipótesis basada en la evidencia de un retraso en la activación del subepicardio del VD o del TSVD (presencia de PT y fraccionamientos); y (3) la más reciente, la hipótesis de corrientes «electrotónicas», basada en una mezcla de alteraciones funcionales y estructurales que condiciona una falla en la excitación del epicardio del VD o del TSVD por un desequilibrio entre la corriente y la carga («current-to-load mismatch»). Las hipótesis 2 y 3 implican la presencia de un miocardio con estructura anormal en la región del TSVD. Se han demostrado anormalidades tales como la presencia de tejido adiposo o de colágeno entre las fibras musculares tanto en el VD como en el TSVD de corazones de individuos con el síndrome.
En apoyo de la «teoría 3», recientemente Ten Sande et al.39 realizaron un mapeo endocárdico y epicárdico en pacientes con el síndrome y demostraron: en primer lugar, que tanto la elevación del segmento ST como la presencia de electrogramas fraccionados y el retraso de la activación surgen de la misma zona, del subepicardio del TSVD. En segundo lugar, que cuando se realiza la ablación de las áreas con electrogramas locales anormales (fraccionados) se normaliza el ST (confirmación de estudios previos de otros grupos). En tercer lugar, que el área con potenciales fraccionados aumenta en forma significativa después del bloqueo del canal de sodio con ajmalina. Estos hallazgos indican que se requieren ambas alteraciones, anatómicas y funcionales, para el desarrollo del patrón ECG característico del síndrome.
Existe otra hipótesis, totalmente distinta a lo antes mencionado y que fue expuesta por Elizari et al. en el 200736. Ellos especularon que la patogénesis del síndrome podría estar relacionada con la expresión anormal de las células de la cresta neural durante el desarrollo del corazón. Expusieron de manera detallada cuál es la relación entre la cresta neural y la conformación del TSVD y cómo puede haber una relación entre estas células y las uniones intercelulares que tendrían los sujetos adultos a través de las conexinas. De esta manera se generaría una heterogeneidad de la repolarización ventricular con las consecuencias arriba señaladas.
Estudios de imagenEcocardiograma 2D, angiografía, imágenes de resonancia magnética y tomografía por haz de electronesComo se documenta en las necropsias de individuos con SBr, la sustitución fibroadiposa interesa principalmente el TSVD, que es una estructura delgada, difícil de investigar para detectar anormalidades histológicas sutiles con todas las técnicas convencionales de imagen. Esta es la razón principal por la cual la mayoría de los pacientes con el verdadero síndrome fueron diagnosticados como portadores de corazones normales por ecocardiografía 2D73. Los datos de anatomía patológica informan que también otras áreas del VD pueden estar involucradas, en particular la pared libre. Por lo general, el VD solo se dilata ligeramente (o nada), pero las anomalías del movimiento de la pared pueden ser detectadas mediante un análisis cuidadoso y detallado con diferentes técnicas de imagen74. Un estudio de imagen por resonancia magnética cardiaco anormal fue informado primero por D’Onofrio et al.75, quienes documentaron el reemplazo graso del TSVD en un paciente con un verdadero síndrome. Los informes posteriores fueron controversiales76. De acuerdo con Santangeli et al.77 la captación tardía de gadolinio es significativamente menos sensible que el mapeo electroanatómico en la identificación de los sustratos de la miocardiopatía del VD, en particular a nivel del TSVD (sobre todo en los casos con cicatrices pequeñas).
Hay escasos estudios de tomografía por haz de electrones o angiografía con radionúclidos para estudiar el VD en el SBr; con ambos métodos se detectaron anomalías del VD y del TSVD78.
Angiografía cardiacaA pesar del empleo extendido del estudio electrofisiológico invasivo en estos sujetos, se dispone de datos limitados sobre el estudio angiográfico del ventrículo izquierdo y del VD, donde se han descrito algunas anomalías menores1,2, recientemente confirmadas por Duthoit et al.79. Resulta discutible un estudio de angiografía derecha normal vs. anormal en los casos con anomalías muy localizadas. Entonces, la experiencia del operador y los protocolos empleados tienen un papel relevante en el estudio del VD.
En el informe de Remme et al.15 el VD fue normal en pacientes con un diagnóstico posterior de DAVD en la necropsia. Como indican colegas de alta experiencia80, una vista única posteroanterior tiene una precisión limitada en la detección de anomalías menores del VD y se necesitan múltiples vistas para un diagnóstico correcto. Una correlación entre cualquier anomalía detectada y el mapeo electroanatómico aumenta la precisión.
Estudios genéticosCuando la enfermedad se presenta en forma hereditaria es una entidad autosómica dominante pero con expresión variable, que afecta principalmente a los hombres y posiblemente más frecuente en Asia3. Después del trabajo original de Chen et al.10 y de Corrado et al.81 se han descrito múltiples mutaciones en el canal de sodio SCN5A82,83, lo que ha llevado a clasificar el síndrome entre las enfermedades de canales iónicos, sin datos claros basados en la evidencia. De acuerdo con los expertos, las mutaciones genéticas de los canales iónicos aún no forman parte de los criterios diagnósticos del síndrome. Según Hoogendick84, y a pesar de la presión que puedan ejercer muchos investigadores básicos (incluyendo biólogos, genetistas, cardiólogos) e incluso la misma industria de los estudios genéticos moleculares, el papel causal exclusivo de las mutaciones genéticas en la fisiopatología del síndrome continúa siendo una hipótesis. Este es claramente familiar en un número importante de pacientes, pero un patrón mendeliano autosómico dominante de transmisión no puede explicar el síndrome en su totalidad. Hasta la fecha, más de 300 mutaciones en 12 genes se han asociado con el síndrome84,85. La mayoría de estas mutaciones reducen la corriente cardiaca de sodio INA y se localizan en el SCN5A10. Otras mutaciones reducen la corriente de calcio tipo L (ICaL) o implican las corrientes de potasio KCNE3 o KCNJ8 y HCN4. Lo que se pone de manifiesto a partir de los estudios publicados es que los pacientes con mutaciones relativas al canal SCN5A, en especial los más jóvenes, tienen mayores defectos de la propagación del impulso (intervalos PR y HV más prolongados y complejos QRS más anchos), mayor fragmentación del QRS y PT. Solo los pacientes y sus familiares con una proteína truncada pueden tener un fenotipo más severo. Curiosamente, el paciente sometido a estudio de necropsia en nuestro informe inicial2,8 tenía anormalidades tanto de DAVD como de SCN5A en su familia. Se ha informado una familia con una nueva mutación en SCN5A (R814Q) que se asoció con un segmento ST en domo (tipo 1) pero también presencia de ondas épsilon y TV monomórfica sostenida con imagen de BRI así como anormalidades estructurales en el VD43. Probst et al. y Marian85,86 demostraron que las anormalidades del SCN5A no son responsables del patrón electrocardiográfico. Ackerman ha propuesto recientemente detener la búsqueda del genotipo en sujetos con el fenotipo del síndrome87.
Como la enfermedad tiene una histología similar a la de la DAVD, se han buscado anomalías genéticas comunes aunque con pobres resultados, pero esto no excluye que estas 2 entidades, similares pero no idénticas, puedan ser producidas por diferentes genes88. Hay informes que proponen que las anormalidades genéticas de la placofilina (PKP2) y la desmoplaquina puedan estar relacionadas con la enfermedad89–91 y seguramente en un futuro cercano habrá nuevos descubrimientos a este respecto92–94.
ConclusiónEn tiempos recientes se ha propuesto una fisiopatología integral para entender las manifestaciones electrocardiográficas del SBr. Los estudios han confirmado los primeros datos científicos de 3 décadas atrás. El patrón ECG resulta inducido por una enfermedad estructural del sistema de conducción, localizada en la rama derecha del haz de His y en el TSVD. El patrón de elevación del segmento ST principalmente se debe a un trastorno de la conducción localizado en dicho tracto (posiblemente asociado a la falta de coincidencia de corriente a la carga [current-to-load-mismatch]), debido a una sustitución fibroadiposa de esta compleja estructura. El resto del VD por lo general tiene una función normal aunque con frecuencia se detecten anomalías estructurales de su pared libre y del sistema de conducción en estudios necrópsicos detallados. El final de esta historia todavía está lejos porque no se han aclarado por completo todas las anomalías genéticas y aún falta evidencia de su fisiopatología.
FinanciaciónNo se recibió patrocinio de ningún tipo para llevar a cabo este artículo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.