





La telangiectasia hemorrágica hereditaria (THH) o enfermedad de Rendu-Osler-Weber, es una condición poco frecuente caracterizada por múltiples telangiectasias y malformaciones arteriovenosas que pueden llevar a episodios repetitivos de epistaxis y otros tipos de sangrado. Las manifestaciones clínicas en la THH son causadas por el desarrollo de la vasculatura anormal (incluyendo telangiectasias, malformaciones arteriovenosas y aneurismas). Se han identificado mutaciones de herencia autosómico dominante en al menos 3 genes: endoglina (ENG), ACVRL1 (HHT2) y SMAD4, y las mutaciones en los primeros 2 explican el 85% de los casos1.
La presentación clínica consiste en epistaxis de repetición, así como en telangiectasias mucocutáneas y malformaciones arteriovenosas viscerales. El diagnóstico se basa en los criterios de Curaçao2 (tabla 1). Si bien las alteraciones mucocutáneas y las malformaciones arteriovenosas son las manifestaciones distintivas del síndrome de Rendu-Osler-Weber, también se ha reportado la asociación de hipertensión pulmonar (HP) como una complicación importante. Reportamos el caso de una paciente con síndrome de Rendu-Osler-Weber e HP.
Criterios de Curaçao para el diagnóstico de telangiectasia hemorrágica hereditaria
| Criterio | Definición |
|---|---|
| Epistaxis | Deben ser espontáneas y recurrentes |
| Telangiectasias | Múltiples en sitios característicos: labios, cavidad oral, dedos, nariz |
| Lesiones viscerales | – Telangiectasias gastrointestinales (con o sin sangrado) – Malformaciones arteriovenosas pulmonares – Malformaciones arteriovenosas hepáticas – Malformaciones arteriovenosas cerebrales – Malformaciones arteriovenosas espinales |
| Historia familiar | Descendente de un primer grado diagnosticado de THH según estos criterios |
THH: telangiectasia hemorrágica hereditaria.
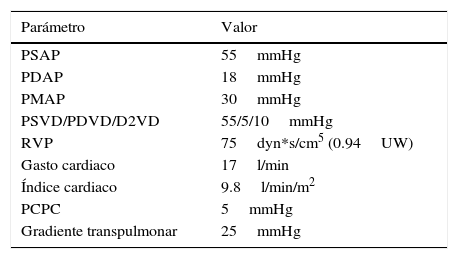
Mujer de 42 años que acudió a consulta a nuestro instituto con historia de 2 años de disnea de grandes esfuerzos y fatiga. Entre sus antecedentes patológicos se incluía un embarazo complicado por preeclampsia a los 40 años de edad y sangrado uterino anómalo de repetición. Negaba consumo de medicamentos o drogas. Al interrogatorio dirigido destacaba la presencia de epistaxis recurrente desde los 23 años de edad. El examen físico demostró la presencia de numerosas telangiectasias en cavidad oral y en miembros pélvicos (fig. 1). El examen del precordio con 2P reforzado y soplo holosistólico grado II/IV en foco tricúspide. El electrocardiograma en ritmo sinusal, con ondas P pulmonares y crecimiento del ventrículo derecho. Un ecocardiograma transtorácico mostró crecimiento de cavidades derechas, presión sistólica arterial pulmonar de 52mmHg e insuficiencia tricuspídea moderada a severa. Se realizó una tomografía toracoabdominal que demostró numerosas malformaciones arteriovenosas a nivel de cuello, hígado y bazo (fig. 2). Se realizó el diagnóstico de síndrome de Rendu-Osler-Weber. Ante la sospecha de HP, se realizó un cateterismo derecho que demostró los resultados citados en la tabla 2. Destaca la presencia de gasto cardiaco elevado (17l/min; IC 9.8l/min) y de presión sistólica de la arteria pulmonar de 55mmHg. Se inició tratamiento diurético con adecuada respuesta y mejoría sintomática. En el seguimiento a 3 meses, la paciente refería asintomática y en buena clase funcional.

.")
Parámetros hemodinámicos relevantes
| Parámetro | Valor |
|---|---|
| PSAP | 55mmHg |
| PDAP | 18mmHg |
| PMAP | 30mmHg |
| PSVD/PDVD/D2VD | 55/5/10mmHg |
| RVP | 75dyn*s/cm5 (0.94UW) |
| Gasto cardiaco | 17l/min |
| Índice cardiaco | 9.8l/min/m2 |
| PCPC | 5mmHg |
| Gradiente transpulmonar | 25mmHg |
D2VD: diámetro telediastólico del ventrículo derecho; PCPC: presión capilar pulmonar en cuña; PDAP: presión diastólica de la arteria pulmonar; PDVD: presión telediastólica del ventrículo derecho; PMAP: presión media de la arteria pulmonar; PSAP: presión sistólica de la arteria pulmonar; PSVD: presión sistólica del ventrículo derecho; RVP: resistencias vasculares pulmonares; UW: unidades Wood.
Varias series han reportado una asociación entre el THH y la HP. En un estudio de Ginon et al.2 se incluyeron 55 pacientes con THH, malformaciones AV hepáticas y disnea o cualquier síntoma cardiológico a quienes se les realizó ecocardiograma transtorácico para evaluar la causa de su disnea. En esta serie, el 25% de los pacientes presentaron una PSAP mayor a 40mmHg. En otra serie de la Clínica Mayo, el 13% de los pacientes con THH podrían mostrar cierto grado de HP utilizando el ecocardiograma como herramienta de detección3.
Se han descrito 2 mecanismos causales de HP en THH: 1) mediante remodelado vascular con aumento en las resistencias vasculares pulmonares, y 2) mediante hiperflujo sistémico y aumento del índice cardiaco (insuficiencia cardiaca de alto gasto). El primer grupo comprende a aquellos pacientes con hipertensión arterial pulmonar clasificados dentro del grupo 1 de la Organización Mundial de la Salud (en específico dentro del subgrupo 1.2.2, hipertensión arterial pulmonar hereditaria asociada a mutaciones de ALK-1 o ENG). En esta entidad se encuentran aumentadas las resistencias vasculares pulmonares y existe un remodelado arterial patológicamente indistinguible de la hipertensión arterial pulmonar tipo 1.
El segundo grupo de pacientes comprende a aquellos con resistencias vasculares pulmonares normales y con la presencia de gasto cardiaco elevado. No existe un subgrupo específico en el consenso de Niza (2013) para clasificar a estos pacientes. Algunas situaciones similares donde existe alto gasto cardiaco incluyen a pacientes con esquistosomiasis (1.4.5), con anemia hemolítica crónica (1.4.6) o a pacientes con desórdenes hematológicos como síndromes mieloproliferativos (donde se ha identificado alto gasto cardiaco e HP, con resistencias pulmonares normales). Sin embargo, al momento de la redacción, la HP asociada a gasto cardiaco elevado por malformaciones AV sistémicas en pacientes con Rendu-Osler-Weber no se incluye dentro de la clasificación de la OMS para HP.
La mayoría de los casos asociados a estados de alto flujo pulmonar se asocian a su vez a malformaciones AV hepáticas y sistémicas4. En el segundo escenario, la hipertensión arterial pulmonar se asocia a mutaciones en las proteínas ALK-1 y ENG (receptor similar a la activina cinasa-1 y ENG)5. El diagnóstico diferencial entre ambos mecanismos fisiopatológicos se realiza mediante parámetros hemodinámicos del cateterismo derecho. En el caso de nuestra paciente, los parámetros hemodinámicos son compatibles con un estado de alto gasto cardiaco (17l/min) que lleva a la HP en presencia vasculatura pulmonar (arterial y venosa) normal y corazón izquierdo normal (estado de alto flujo pulmonar).
La experiencia en el tratamiento médico de los pacientes con HP en el contexto de la THH se limita a reportes de casos. No existen ensayos clínicos aleatorios, y es improbable que sean realizados. Los pacientes con HP secundaria a insuficiencia cardiaca de alto flujo pueden beneficiarse del tratamiento con restricción de sal y diuréticos6; la embolización transarterial de malformaciones AV hepáticas se ha propuesto como una opción, pero puede causar complicaciones como infarto o sepsis, por lo que en general no se utiliza. El trasplante hepático podría ser una opción terapéutica en casos especiales7. El tratamiento dirigido a la THH (bevacizumab, un anticuerpo monoclonal que inhibe el factor de crecimiento vascular endotelial) podría revertir las lesiones hepáticas y reducir el gasto cardiaco en pacientes con importante carga de malformaciones AV hepáticas y sistémicas8.
En cuanto a los pacientes con hipertensión arterial pulmonar, el tratamiento estándar incluyendo bosentan y tadalafil, se ha extrapolado con algunos resultados positivos9. Sin embargo es importante recordar que estas terapias pueden incrementar el riesgo de sangrado, el cual es alto en los pacientes con THH.
Sabbà et al.10 demostraron una disminución aproximada de 7 años en la expectativa de vida en pacientes con HP y THH. El estudio de la Clínica Mayo falló en alcanzar significación estadística, pero si mostró una tendencia de la HP como factor predictivo de pronóstico adverso y sobrevida disminuida6. Es por esto que resulta importante aumentar el grado de sospecha clínica, y clarificar la importancia del cateterismo derecho para confirmar el diagnóstico e identificar el mecanismo fisiopatológico.
Se debe mantener una alta sospecha clínica sobre el diagnóstico de la HP en aquellos pacientes con THH que presenten disnea, o en quienes un ecocardiograma identifique incremento en la presión sistólica del ventrículo derecho o incremento en la presión de la aurícula derecha.
FinanciaciónNo se recibió patrocinio de ningún tipo para llevar a cabo este artículo.