



La enfermedad tromboembólica venosa es de tipo complejo. Es consecuencia de la interacción de factores ambientales y genéticos que muchas veces se reflejan en los llamados fenotipos intermediarios. Estos se asocian por definición al riesgo de trombosis y por ello tienen una gran utilidad tanto diagnóstica como en la búsqueda de los factores causales últimos (ambientales y genéticos). Las investigaciones actuales van encaminadas a responder por qué se origina un trombo mediante la búsqueda de nuevos mecanismos que se añaden a la tríada descrita por Virchow. Los estudios de genome wide asociation study (GWAS) y las técnicas de secuenciación masiva han permitido identificar nuevas variantes genéticas. A estas técnicas se les suman nuevos campos en desarrollo como la epigenética, la transcriptómica y el estudio del metaboloma. El procesamiento de la inmensa información mediante los algoritmos adecuados y modelos matemáticos y bioinformáticos multiplicará la capacidad para responder cuestiones sobre el espectro mutacional de la trombosis y estimar el riesgo individual de padecerla. El área de investigación de mayor valor clínico aplicado en trombofilia debe ir encaminada al desarrollo de modelos predictivos que permitan estimar el riesgo individual de trombosis o de recidiva, si es el caso.
Thrombosis is a complex human disease. It is the result of the interaction between the environment and genes. This interaction is manifested by «intermediary phenotypes». These intermediary phenotypes are useful for estimating the risk of developing the disease. The targets of current investigations are attempting to identify new mechanisms that underlie the classic Virchow's triad. Modern methodologies, such as genome wide association studies (GWAS) and high-throughput genotyping technology (massive sequencing techniques) are being used to identify genetic variants. The fields of epigenetics, transcriptomics and metabolomics are being developed to complement these studies. The vast amount of data to explain the environmental and genetic factors that predispose for the disease require informatic techniques, such as mathematical and bio-computer models to estimate the individual risk of thrombosis. The research area with the highest clinical application is the development of predictive models to estimate the individual risk of thrombosis.
El conocimiento actual de la etiología de la trombosis se basa en la primera descripción realizada a mediados del siglo xix por el patólogo Virchow. Virchow postuló que las principales causas de trombosis se resumían en cambios en el vaso sanguíneo, el flujo y en los componentes de la sangre. Estas premisas actualmente siguen siendo válidas1. La trombofilia puede considerarse un estado latente de la coagulación en el que la resistencia a la trombosis se encuentra disminuida, aumentando la posibilidad de formación de trombos arteriales y venosos2. Los mecanismos que anticipó Virchow en un intento de explicar la trombosis, son el resultado de la interacción de factores de riesgo ambientales (o adquiridos) y genéticos.
En 1996, un comité de expertos de la OMS y la ISTH definió la trombofilia hereditaria como una tendencia genéticamente determinada al tromboembolismo venoso (TEV). Anomalías dominantes, en algunos casos, y combinaciones de defectos más leves, en otros, pueden causar TEV a edad de inicio temprana, recidivas frecuentes o historia familiar de trombosis. Los rasgos leves pueden ser solo revelados mediante investigación de laboratorio. Todavía no se conocen ni se han comprendido todas las influencias genéticas ni sus interacciones3. Esta definición implicaba que tan solo un subgrupo selecto de pacientes con TEV debía su enfermedad a variantes genéticas (algunas conocidas, como los déficits de antitrombina, proteína S o proteína C, o las mutaciones Factor V Leiden o PT20210A, y otras, todavía por descubrir). En contraposición, la gran mayoría de casos observados en la clínica diaria eran atribuidos a circunstancias ambientales o adquiridas, con muy poca o nula influencia genética.
Pocos años más tarde, se impuso la noción de que cualquier episodio de trombosis es el resultado de la interacción de genes y factores ambientales, obligando a entender la enfermedad tromboembólica venosa como un rasgo complejo. La base genética subyacente a esta complejidad parece ser la parte más importante, explicando entre un 50 y un 60% de la predisposición a la enfermedad4–6. Sin embargo, hasta la fecha, solo hemos conseguido detectar una parte muy pequeña, alrededor del 5%, de esta base genética o heredabilidad7.
Es necesario ampliar el campo de investigación de las causas de trombosis más allá de los clásicos estudios realizados hasta ahora. Principalmente se ha investigado en sujetos con trombosis espontáneas, con características de trombofilia hereditaria: de edad joven, con trombosis recidivante y con historia familiar positiva. Este grupo de pacientes supone un porcentaje muy pequeño de las trombosis observadas en la vida real. Si la base genética de la enfermedad juega un papel importante en cualquier situación clínica, será conveniente incluir todo tipo de pacientes en estudios futuros, por ejemplo, casos de trombosis paraneoplásicas, las trombosis en pacientes de edad avanzada y las asociadas a factores de riesgo adquiridos, como la cirugía u otras comorbilidades. Idealmente, estas investigaciones deberían ofrecer respuestas de utilidad clínica a 3 preguntas importantes: 1.ª) ¿por qué un sujeto concreto sufre una trombosis? 2.ª) ¿cuál es el riesgo individual de sufrir una trombosis tanto en personas asintomáticas (riesgo basal) como en pacientes con trombosis previa (riesgo de recidiva)?, y la pregunta más difícil de responder y que necesita del conocimiento de las respuestas previas: 3.ª) ¿cuándo un sujeto concreto va a presentar una trombosis o una recidiva, si es el caso?
Causas de tromboembolismo venoso. ¿Por qué un paciente ha sufrido una trombosis?En condiciones normales, ante la rotura de un vaso, se origina un trombo como mecanismo defensivo para evitar la pérdida de sangre. Cuando este proceso tiene lugar fuera de este contexto, es cuando nos encontramos con una trombosis patológica.
Utilizando la tríada de Virchow como matriz, podemos añadir nuevos mecanismos que en ausencia de traumatismo vascular ocasionan un trombo patológico. Atendiendo a una reciente revisión de Reitsma8 en la que se describen los factores de riesgo de trombosis más comunes, intentaremos realizar un análisis sintético para su comprensión. Uno de los factores de riesgo a considerar es la hipoxemia. En las zonas valvulares venosas, la concentración de oxígeno es menor, acentuándose en aquellos casos en que las válvulas se encuentran dañadas. La hipoxemia da lugar a unas condiciones proinflamatorias y procoagulantes que implican distintos agentes y mecanismos, entre ellos, las proteínas pro- y anticoagulantes, la vía del factor tisular, la p-selectina, las plaquetas, las micropartículas y la activación de la inmunidad innata (incluyendo monocitos y granulocitos) y adquirida.
- 1.
Las proteínas activadoras e inhibidoras de la coagulación: Como es sabido, los determinantes de riesgo de trombosis mejor estudiados son los componentes de la coagulación plasmática tanto los factores procoagulantes como los inhibidores. Muchas de las variaciones, cuantitativas o cualitativas, en la concentración plasmática se explican por anomalías genéticas como el déficit de antitrombina, de proteína C, de proteína S, la mutación del factor V Leiden, la mutación de la protrombina y el grupo sanguíneo no O. Aunque estas alteraciones génicas patofisiológicamente puedan explicar la tendencia a la coagulación, es necesaria la interacción con otros mecanismos relacionados con el estilo de vida y los factores ambientales para que en un momento determinado alteren el equilibrio entre los factores protrombóticos y los inhibidores.
- 2.
El papel de la inmunidad: Como consecuencia de los procesos inflamatorios e infecciosos que tienen lugar en la zona donde se originará el trombo, se produce la activación de la inmunidad innata involucrando la activación del endotelio, de los granulocitos y los monocitos. Los granulocitos neutrófilos activados pueden liberar material intranuclear formando redes extracelulares o neutrophil extracellular traps (NET). Las NET son una barrera física inicialmente destinada a evitar la diseminación de microorganismos, pero también suponen un andamiaje molecular sobre el que se desencadenan los procesos de coagulación y formación de fibrina. En un artículo publicado por van Montfoort et al.9, se describe la correlación entre el desarrollo de las NET (como consecuencia de la activación de los neutrófilos) y el riesgo de TEV. Una forma indirecta de objetivar la activación de los neutrófilos es mediante la medición de los complejos elastasa/alfa-1-antitripsina y los niveles de nucleosomas circulantes. Se ha visto que los nucleosomas no solo forman parte de las NET sino que también se detectan en procesos que comportan una destrucción celular, principalmente cuando implican la muerte de células endoteliales. De hecho, existen datos epidemiológicos que relacionan el aumento del riesgo de trombosis en el contexto de enfermedades infecciosas10.
- 3.
Las plaquetas: Hasta ahora se había prestado escasa atención a las plaquetas como agentes relevantes en la patogénesis del TEV. Actualmente, se conoce que las plaquetas activadas son catalizadoras importantes de la activación de la protrombina (tanto a través de la vía intrínseca como de la vía extrínseca) para generar trombina y, finalmente, la red de fibrina11. También se han visto involucrados en estos mecanismos los receptores plaquetarios, como la glucoproteína VI (receptor para el colágeno), que interviene en los procesos de agregación y adhesión celular y que posee variantes genéticas asociadas al riesgo de TEV12. En apoyo al papel de las plaquetas en la etiología del TEV, cabe mencionar que los fármacos inhibidores de la función plaquetaria, como los antiagregantes, disminuyen la incidencia de TEV tanto primario como recurrente13. Esta afirmación se apoya en los resultados que ofrecieron los estudios ASPIRE14 y WAFASA15 en los que se reclutaron pacientes que habían padecido un TEV profundo proximal espontáneo. El objetivo primario era evaluar el riesgo de recurrencia de TEV en pacientes tras haber finalizado el tratamiento con fármacos antivitamina K. Un grupo recibió dosis bajas de aspirina frente el otro que recibió placebo. Ambos estudios demostraron una disminución en el riego de recurrencia de TEV y de eventos vasculares mayores (accidentes vasculares cerebrales, infartos agudos de miocardio) sin aumentar significativamente el riesgo de sangrado en el grupo tratado con antiagregantes.
Otro aspecto que cabe mencionar es el papel de los polifosfatos contenidos en los gránulos densos plaquetarios. Los polifosfatos son polímeros sencillos constituidos por fosfatos inorgánicos unidos entre sí. Su función es ser fuente de fosfatos para la síntesis de las moléculas de ATP en casos de mayor demanda energética. En el plasma humano, la vida media de los polifosfatos es de 1,5 a 2 h ya que están sometidos a la degradación de las fosfatasas circulantes. Cuando se produce la activación plaquetaria, se libera el contenido de sus gránulos. Los polifosfatos intervienen en toda la vía de la coagulación. Por una parte, como son polímeros aniónicos, se unen a proteínas que iniciarían la fase de contacto de la coagulación o vía intrínseca. También aceleran la activación del factor v mediante la activación del factor x y la generación de trombina. Cuando el factor xa está unido al cofactor va, es indemne a la acción de la proteína inhibidora del factor tisular (TFPI), esta proteína no degrada el factor tisular y por lo tanto se favorece el proceso coagulativo. El factor xi se puede activar fundamentalmente por 3 vías: mediante el factor xiia, por feedback positivo del factor xia y mediante la trombina. Estas 2 últimas son muy lentas fisiológicamente, viéndose aceleradas en condiciones en que aumentan los niveles de polifosfato. También contribuyen en la fase final de la cascada de la coagulación mediante la generación de mallas de fibrina de mayor longitud y grosor siendo más resistentes a la fibrinólisis16.
- 4.
Las micropartículas (MP): Las MP son vesículas procedentes de la superficie celular con distintos orígenes. La mayoría (80%) se forman en la superficie de las plaquetas, pero también podemos encontrar procedentes de monocitos y de células endoteliales. Las proteínas que conforman su superficie reflejan su procedencia. Entre estas proteínas cabe destacar una de ellas: el factor tisular. Se ha establecido una correlación entre la expresión de factor tisular en la superficie de algunas MP y el riesgo de TEV por su actividad procoagulante capaz de activar la vía extrínseca17.
Los anteriores mecanismos, entre otros, son el resultado de una conjunción de factores causales algunos de ellos presentes en el ambiente del sujeto y otros pertenecientes a su base genética. Queda claro que cualquier TEV es de naturaleza multifactorial, es decir, resulta de la interacción de múltiples factores ambientales y genéticos. La identificación de estos factores nos permite entender, aunque parcialmente, los casos que observamos en la clínica diaria. Sin embargo, la comprensión de cada caso es todavía muy insuficiente por 3 motivos: porque aún desconocemos la mayor parte de determinantes genéticos, porque seguramente quedan factores ambientales desencadenantes por descubrir, y porque a pesar de identificar ciertos factores de riesgo, desconocemos cómo interactúan entre sí, o el valor relativo de cada uno de ellos en un paciente concreto.
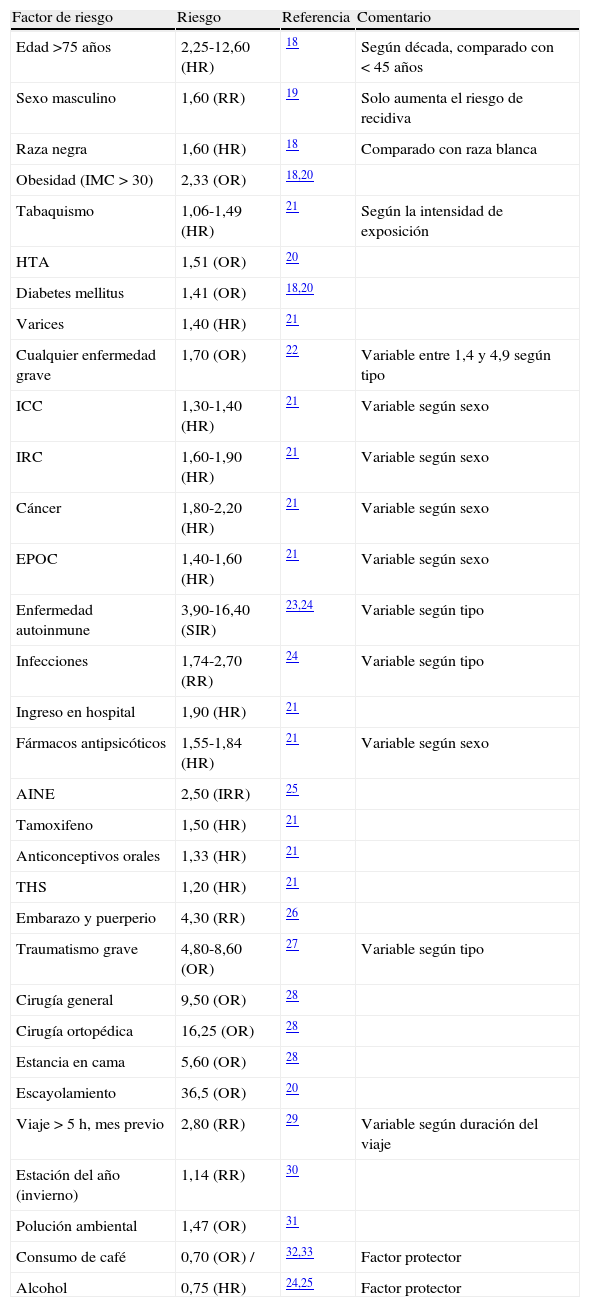
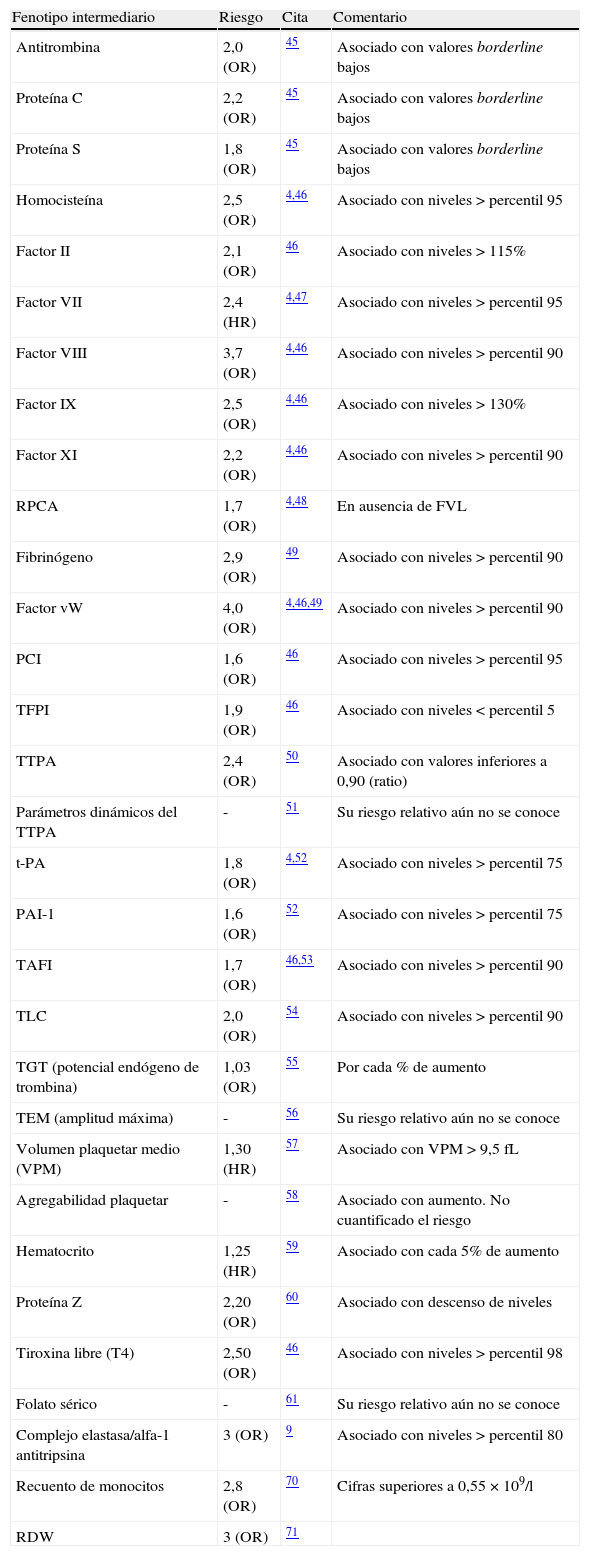
Cualesquiera que sean las variantes genéticas de riesgo de trombosis que un paciente posea, o las circunstancias ambientales que le influyan, nunca observamos una afectación clínica continua. Incluso en los casos más llamativos de trombofilia, en la mayoría de pacientes, la trombosis sucede en episodios separados a menudo por periodos asintomáticos prolongados. Esta discontinuidad sugiere la necesidad de factores desencadenantes para cada episodio, tal vez estímulos directos, el deterioro temporal de resistencia intrínseca a la trombosis, o alguna combinación de estos. Este característico modo de presentación en forma de sucesos separados en el tiempo, subraya la importancia de la interacción con el ambiente. Para explicar cualquier episodio de TEV disponemos actualmente de una relación numerosa de factores de riesgo ambiental (tabla 1) y otra de riesgo genético (tabla 2). También se han identificado múltiples parámetros de laboratorio, técnicamente considerados como «fenotipos intermediarios» que se asocian con el riesgo de trombosis (tabla 3). Todos estos fenotipos intermediarios también son «complejos», es decir, son el resultado de la interacción de genes y ambiente. Su vinculación con la enfermedad puede ser utilizada para 3 propósitos de gran interés: a) explicar los casos clínicos, b) formar parte de los modelos de predicción (ver más adelante) y también c) en la búsqueda de factores genéticos aún desconocidos.
Factores adquiridos o ambientales asociados con el riesgo de TEV
| Factor de riesgo | Riesgo | Referencia | Comentario |
| Edad >75 años | 2,25-12,60 (HR) | 18 | Según década, comparado con < 45 años |
| Sexo masculino | 1,60 (RR) | 19 | Solo aumenta el riesgo de recidiva |
| Raza negra | 1,60 (HR) | 18 | Comparado con raza blanca |
| Obesidad (IMC > 30) | 2,33 (OR) | 18,20 | |
| Tabaquismo | 1,06-1,49 (HR) | 21 | Según la intensidad de exposición |
| HTA | 1,51 (OR) | 20 | |
| Diabetes mellitus | 1,41 (OR) | 18,20 | |
| Varices | 1,40 (HR) | 21 | |
| Cualquier enfermedad grave | 1,70 (OR) | 22 | Variable entre 1,4 y 4,9 según tipo |
| ICC | 1,30-1,40 (HR) | 21 | Variable según sexo |
| IRC | 1,60-1,90 (HR) | 21 | Variable según sexo |
| Cáncer | 1,80-2,20 (HR) | 21 | Variable según sexo |
| EPOC | 1,40-1,60 (HR) | 21 | Variable según sexo |
| Enfermedad autoinmune | 3,90-16,40 (SIR) | 23,24 | Variable según tipo |
| Infecciones | 1,74-2,70 (RR) | 24 | Variable según tipo |
| Ingreso en hospital | 1,90 (HR) | 21 | |
| Fármacos antipsicóticos | 1,55-1,84 (HR) | 21 | Variable según sexo |
| AINE | 2,50 (IRR) | 25 | |
| Tamoxifeno | 1,50 (HR) | 21 | |
| Anticonceptivos orales | 1,33 (HR) | 21 | |
| THS | 1,20 (HR) | 21 | |
| Embarazo y puerperio | 4,30 (RR) | 26 | |
| Traumatismo grave | 4,80-8,60 (OR) | 27 | Variable según tipo |
| Cirugía general | 9,50 (OR) | 28 | |
| Cirugía ortopédica | 16,25 (OR) | 28 | |
| Estancia en cama | 5,60 (OR) | 28 | |
| Escayolamiento | 36,5 (OR) | 20 | |
| Viaje > 5 h, mes previo | 2,80 (RR) | 29 | Variable según duración del viaje |
| Estación del año (invierno) | 1,14 (RR) | 30 | |
| Polución ambiental | 1,47 (OR) | 31 | |
| Consumo de café | 0,70 (OR) / | 32,33 | Factor protector |
| Alcohol | 0,75 (HR) | 24,25 | Factor protector |
AINE: antiinflamatorios no esteroideos; EPOC: enfermedad pulmonar obstructiva crónica; HR: hazard ratio; HTA: hipertensión arterial; ICC: insuficiencia cardiaca congestiva; IMC: índice de masa corporal; IRC: insuficiencia renal crónica; IRR: incidence rate ratio; OR: odds ratio; RR: relative risk; SIR: standardised incidence ratio; THS: terapia hormonal sustitutiva.
Factores genéticos asociados con el riesgo de TEV
| Gen o variante genética | RR | Cita | Comentario |
| SERPINC1 (antitrombina) | 10 | 7 | Más de 130 mutaciones privadas con déficit AT en plasma |
| AT Cambridge II (Ala384Ser) | 10 | 34 | Muy difícil de detectar en plasma |
| SERPINC1 rs2227589 | 1,30 | 7 | |
| PROC (proteína C) | 8 | 7 | Más de 160 mutaciones privadas con déficit de PC |
| PROCR (receptor endotelial) rs867186 | 1,22 | 7 | |
| PROS1 (proteína S) | 8 | 7 | Más de 200 mutaciones privadas con déficit de PS |
| F5 rs6025 (FV Leiden) | 5 | 7 | |
| F5 rs4524 | 1,21 | 7 | Independiente de FVL (no están en desequilibrio de ligam.) |
| F5 Cambridge (Arg306Thr) | - | 35 | Provoca RPCA. Su riesgo relativo no se conoce |
| F5 Hong-Kong (Arg306Gly) | - | 36 | Provoca RPCA. Su riesgo relativo no se conoce |
| F2 rs1799963 (PT20210A) | 3 | 7 | |
| ABO rs8176719 y rs8176750 | 2-4 | 7 | Corresponde con los portadores B y A1 del grupo ABO |
| VWF rs1063856-C | 1,15 | 7 | Aumenta los niveles de FvW y el riesgo de TEV |
| STAB2 rs4981021 | 1,29 | 7 | |
| TC2N rs1884841 | 1,22 | 7 | |
| STXBP5 rs1039084-G | 0,78 | 7 | Factor protector |
| FGG rs2066865 | 1,47 | 7 | Este SNP disminuye la relación γA/γ’ y aumenta riesgo TEV |
| F11 rs2036914 y rs2289252 | 1,35 | 7 | Estos SNP modulan los niveles de FXI en plasma |
| GP6 rs1613662 | 1,15 | 7 | |
| HIVEP1 rs169713-C | 1,20 | 7 | |
| KNG1 rs710446 | 1,19 | 7 | Modula FXI, acorta el TTPA y aumenta riesgo de TEV |
| F12 rs1801020 (FXII C46T) | 5 | 37 | El riesgo se asocia a los homocigotos T/T |
| F13A1 rs5985 (FXIII Val34Leu) | 0,85 | 38 | Factor protector asociado al genotipo Leu/leu |
| SERPINA10 rs2232698 | 3,30 | 39 | SNP en el gen inhibidor de proteasa dependiente de Prot. Z |
| Trombomodulina (THBD) rs1042579 | 0,72 | 40 | Relacionado con el aumento de la actividad de la Prot. C |
AT: Antitrombina; RR: riesgo relativo; RPCA: resistencia a la proteína C activada; PC: Proteína C; PS: Proteína S; FXI: Factor onze; SNP: single nucleotide polymorphism; TEV: tromboemblolismo venoso; TPA: tiempo de tromboplastina parcial activado; OR: odds ratio.
Otras variantes genéticas potencialmente relacionadas con el riesgo de TEV (pendientes de confirmación):
Soria et al.41: ANX5 haplotipo M2 (OR 1,90); APOA4 Gln360His (OR 0,34); BAI3 rs9363864 (OR 0,58); CYP4V2 rs13146272 (OR 1.20); SELE (gen de E-selectina) Leu554Phe (OR 0,39); F2 A19911G (OR 1,43 para G/G); F9 rs4149755 (OR 1.50); F10 rs69333 (OR 0,80); FGA rs6050 (OR 1,40) y rs2070006 (OR 1,25); FGB rs1800788 (OR 1,30); IL1RN rs2232354 (OR 3,90); IL4R Ile50Val (OR 0,66); LPL Asn291Ser (OR 3,09); SERPINA 1 (gen del PAI-1) rs2267667 (OR 1,26); KLKB1 rs3087505 (OR 1,27); PROC rs5937 (OR 1,37) y rs2069915 (OR 0,78); PROCR G4678C (OR 0,50); CPB2 (gen del TAFI) rs17844078 (OR 0,52); TFPI rs2192824 (OR 1,25); TNFSF4 −921C/T (OR 1,86).
De Haan et al.42: PROC rs1799809 (OR 1,17); PROCR rs2069951 (OR 1,30); F2 rs3136516 (OR 1,19); F5 rs1800595 (OR 1,18); F8 rs1800291 (OR 1,13); NAT8B rs2001490 (OR 1,10); F13B rs6003 (OR 1,09); RGS7 rs670659 (OR 1,09); F9 rs6048 (OR 1,08); F11 rs3822057 (OR 1,06); NR1I2 rs3742264 (OR 1,05); F3 1208indel (OR 1,06).
Gretarsdottir et al.43: DAB2IP rs7025486 (OR: 1,2).
Kondkar et al.44: VAMP/8 endobrevin rs101.
Fenotipos sanguíneos (intermediarios) asociados con el riesgo de TEV
| Fenotipo intermediario | Riesgo | Cita | Comentario |
| Antitrombina | 2,0 (OR) | 45 | Asociado con valores borderline bajos |
| Proteína C | 2,2 (OR) | 45 | Asociado con valores borderline bajos |
| Proteína S | 1,8 (OR) | 45 | Asociado con valores borderline bajos |
| Homocisteína | 2,5 (OR) | 4,46 | Asociado con niveles > percentil 95 |
| Factor II | 2,1 (OR) | 46 | Asociado con niveles > 115% |
| Factor VII | 2,4 (HR) | 4,47 | Asociado con niveles > percentil 95 |
| Factor VIII | 3,7 (OR) | 4,46 | Asociado con niveles > percentil 90 |
| Factor IX | 2,5 (OR) | 4,46 | Asociado con niveles > 130% |
| Factor XI | 2,2 (OR) | 4,46 | Asociado con niveles > percentil 90 |
| RPCA | 1,7 (OR) | 4,48 | En ausencia de FVL |
| Fibrinógeno | 2,9 (OR) | 49 | Asociado con niveles > percentil 90 |
| Factor vW | 4,0 (OR) | 4,46,49 | Asociado con niveles > percentil 90 |
| PCI | 1,6 (OR) | 46 | Asociado con niveles > percentil 95 |
| TFPI | 1,9 (OR) | 46 | Asociado con niveles < percentil 5 |
| TTPA | 2,4 (OR) | 50 | Asociado con valores inferiores a 0,90 (ratio) |
| Parámetros dinámicos del TTPA | - | 51 | Su riesgo relativo aún no se conoce |
| t-PA | 1,8 (OR) | 4,52 | Asociado con niveles > percentil 75 |
| PAI-1 | 1,6 (OR) | 52 | Asociado con niveles > percentil 75 |
| TAFI | 1,7 (OR) | 46,53 | Asociado con niveles > percentil 90 |
| TLC | 2,0 (OR) | 54 | Asociado con niveles > percentil 90 |
| TGT (potencial endógeno de trombina) | 1,03 (OR) | 55 | Por cada % de aumento |
| TEM (amplitud máxima) | - | 56 | Su riesgo relativo aún no se conoce |
| Volumen plaquetar medio (VPM) | 1,30 (HR) | 57 | Asociado con VPM > 9,5 fL |
| Agregabilidad plaquetar | - | 58 | Asociado con aumento. No cuantificado el riesgo |
| Hematocrito | 1,25 (HR) | 59 | Asociado con cada 5% de aumento |
| Proteína Z | 2,20 (OR) | 60 | Asociado con descenso de niveles |
| Tiroxina libre (T4) | 2,50 (OR) | 46 | Asociado con niveles > percentil 98 |
| Folato sérico | - | 61 | Su riesgo relativo aún no se conoce |
| Complejo elastasa/alfa-1 antitripsina | 3 (OR) | 9 | Asociado con niveles > percentil 80 |
| Recuento de monocitos | 2,8 (OR) | 70 | Cifras superiores a 0,55 × 109/l |
| RDW | 3 (OR) | 71 |
HR: hazard ratio; OR: odds ratio; PAI-1: inhibidor de los activadores del plasminógeno; PCI: inhibidor de la proteína C; RDW: amplitud de distribución eritrocitaria; RPCA: resistencia a la proteína C activada; t-PA: activador tisular del plasminógeno; TAFI: inhibidor de la fibrinólisis activable por trombina; TEM: tromboelastometría; TFPI: inhibidor de la vía del factor tisular; TGT: test de generación de trombina; TLC: tiempo de lisis del coágulo; TTPA: tiempo de tromboplastina parcial activado.
Resulta indudable que la combinación de distintos factores de riesgo (tanto genéticos como ambientales) siempre provoca un riesgo final mucho mayor que la mera suma de riesgos aislados. Por ejemplo, una enfermedad grave provoca un riesgo de 1,7, la inmovilización se asocia con un riesgo de 5,6 (tabla 1) y el aumento de niveles de FVIII con un riesgo de 3,7 (tabla 3). La presencia de los 3 factores en un mismo sujeto le confiere un riesgo de TEV 88 veces superior, según datos del estudio MEGA22.
Futuro de la investigación en trombofiliaA continuación, proponemos 2 campos que consideramos clave en el avance del conocimiento, en el pronóstico y en la prevención de la trombosis.
Completar la base genéticaPuesto que la constitución genética de los individuos explica entre un 50 y un 60% de su predisposición al TEV y las variantes genéticas de riesgo identificadas hasta la actualidad no suponen conjuntamente ni un 10% del riesgo, está claro que un área imprescindible de investigación debe ser el descubrimiento de los factores genéticos aún desconocidos. En la tabla 2 se registran 19 loci genéticos claramente asociados al riesgo y otros 22 potencialmente asociados (pie de tabla, pendientes de confirmación). Sin contar con las mutaciones privadas en los genes SERPINC1, PROC y PROS1, causantes de los déficits patogénicos de antitrombina, proteína C y proteína S, respectivamente. Estos 41 loci identificados albergan al menos 23 variantes de riesgo reconocido y otras 34 todavía por confirmar. La mayoría de estas 57 variantes se han identificado recientemente a través de estudios genómicos de asociación o GWAS. Los GWAS permiten estudiar centenares de miles de variantes genéticas y estimar su posible asociación con el riesgo de TEV mediante diseños epidemiológicos clásicos que comparan casos con controles sanos. Los marcadores genéticos utilizados para localizar los genes causales son del tipo single nucleotide polymorphism (SNP) con una frecuencia alélica relativamente alta (variables comunes).
A pesar de las esperanzas depositadas en estos estudios masivos, los resultados han sido frustrantes. Solo han permitido identificar variantes de alta frecuencia poblacional (presentes en más del 5% de los individuos) pero con un riesgo relativo muy pequeño, menor de 1,50. De ahí que la atención se deba dirigir hacia variantes más raras, como la AT Cambridge34 o la SERPINA10 rs223269839, que suponen unos riesgos relativos muy notables para sus portadores (10 y 3,3 respectivamente). En este sentido se están postulando otros tipos de diseños epidemiológicos, implicando tamaños muestrales mucho menores pero en grupos selectos (por ejemplo, estudios familiares) como fuentes de nuevos descubrimientos. Y, lo que es todavía más importante, estudios que permitan no solamente aumentar nuestra lista de factores de riesgo, sino también mejorar la utilidad y la aplicabilidad clínica de los hallazgos a un nivel individual46.
Sin duda, los avances espectaculares en las técnicas de laboratorio, como la secuenciación de nueva generación (NGS), servirán para identificar variantes raras (o muy raras) responsables de formas familiares. Al unir las técnicas de secuenciación masiva con los algoritmos apropiados, se multiplica la habilidad para responder cuestiones sobre el espectro de mutación de una enfermedad. Otro campo novedoso está relacionado con la epigenética, que es el conjunto de procesos químicos que modifican la actividad del ADN sin alterar la secuencia. Su influencia actual en el TEV es prácticamente desconocida pero deberá ser explorada como candidata a explicar parte de los mecanismos genéticos. En concreto, los procesos de metilación/acetilación del ADN sobre la transcripción génica y los efectos de los micro-ARN sobre la síntesis de proteínas o fenotipos intermediarios involucrados (tabla 3) necesitan ser estudiados para dilucidar su aportación al riesgo de la enfermedad. En relación con estos procesos epigenéticos, los datos provenientes de análisis completos de la transcripción (transcriptómica) y el conocimiento de los elementos químicos que reflejan el estado metabólico de un organismo o metaboloma deberían contribuir a rellenar las actuales lagunas. Para desentrañar completamente la arquitectura genómica de la trombosis, en última instancia, se deberán conocer también otras formas de variabilidad, aparte de los SNP, como las variaciones del número de copias (CNV) y las interacciones gen-gen o gen-ambiente7. Este progreso gigante en la adquisición de datos solo tendrá sentido si, en paralelo, se desarrollan los métodos matemáticos y bioinformáticos imprescindibles para analizar y explotar la descomunal información obtenida.
Modelos predictivosPrecisamente estos métodos de análisis y de procesamiento masivo de información, también serán imprescindibles en el desarrollo de modelos predictivos que permitan estimar el riesgo individual. El perfil de riesgo individual es el conjunto de variables clínicas y biológicas cuya evaluación combinada y ponderada permite estimar de forma cuantitativa y objetiva el riesgo de enfermedad en cada individuo. Idealmente, esto permitiría asignar a cualquier individuo y en cualquier momento, su nivel de riesgo de trombosis permitiendo adoptar las medidas preventivas adecuadas con gran selectividad.
En la actualidad existen múltiples modelos que de una forma bastante simplista (y con poca aplicabilidad individual) se han usado en ámbitos estancos: en pacientes oncológicos61, en modelos predictivos del riesgo de recidiva62, en el embarazo63, en cirugía general64, en cirugía ortopédica65 o en pacientes médicos hospitalizados66. Todos ellos se han basado sobre todo en factores de riesgo clínico, muy específicos de cada situación y sin apenas contar con la base genética del individuo o los datos biológicos que sabemos que se asocian al riesgo de trombosis. En este sentido, resultan mucho más interesantes los esfuerzos para determinar el riesgo basal de un individuo, aunque solo sea con información clínica21. Recientemente ha quedado demostrado que la incorporación de datos genéticos mejora sustancialmente la capacidad predictiva de estos modelos42. Además, se puede también incorporar la información disponible, que es mucha, proporcionada por los fenotipos intermediarios, aumentando más el poder de los modelos67. Otros avances necesarios deben producirse en los métodos matemáticos para abordar la cantidad y complejidad de la información67,68. Por ejemplo, Eichenger et al. propusieron un score de riesgo de recurrencia en pacientes con trombosis espontánea previa. Un año más tarde, Hippsley-Cox21 y Heinneman69 desarrollaron un score para predecir el riesgo de un primer evento de trombosis en individuos asintomáticos. De ellos, solo Heinemann utilizó factores genéticos (2, para ser más exactos) para calcular el riesgo. Más recientemente, de Haan42 aumentó el valor predictivo mediante la adición de más variables genéticas descubiertas en estudios de GWAS. Sin embargo, este trabajo solo consigue explotar la información aportada por 5 variantes genéticas, entre las más de 30 variantes distintas y asociadas independientemente al riesgo de trombosis, que incluye en sus modelos matemáticos. Esto nos invita a plantear la necesidad de utilizar nuevas herramientas matemáticas para responder a las preguntas que nos formulamos hoy en día.
Basta con comprender que las posibilidades combinatorias de los factores de riesgo actualmente aceptados (tablas 1–3) son inmensas, y que el número de perfiles individuales (compuestos por los datos genéticos, epigenéticos, ambientales, plasmáticos y sus interacciones) que definen el riesgo de cada sujeto también es enorme. Para ello, es necesario un buen procesamiento de la información mediante el diseño de estudios (casos y controles, cohortes prospectivas poblacionales) y la utilización de nuevos modelos matemáticos como Multilinear principal component analysis, Independent component analysis, Support vector machines, Partial least squares o Neural network machine learning methods. En el futuro deberíamos poder agrupar los múltiples niveles de riesgo individuales en categorías clínicamente equivalentes. Mediante estos progresos, en años venideros dispondríamos de un modelo unificado para estimar el riesgo de trombosis de cualquier persona y en cualquier situación clínica.
En los últimos años se ha progresado en el descubrimiento de factores de riesgo de trombosis y en los mecanismos por los que estos la producen. Sin embargo, a pesar de tener cada vez más información, seguimos sin poder explicar una parte no menospreciable de los TEV que vemos en la práctica diaria. Esto nos lleva a insistir en la búsqueda de algoritmos que nos permitan fusionar toda la información para definir el riesgo individual de padecer un evento de trombosis.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores no declaran ningún conflicto de interés.
Trabajo realizado con el apoyo de: FIS PI 12/00612 y Red de Investigación Cardiovascular RD12/ 0042/0032.
