




Puntos clave
- •
Los síndromes hemofagocíticos se caracterizan por presentar un defecto en la contracción y una expansión continua e ilimitada de la respuesta inmunitaria.
- •
Las formas primarias de la linfohistiocitosis hemofagocítica familiar (FHLH) tienen una incidencia aproximada de 1/100.000 niños al año.
- •
Además de las formas familiares, la HLH primaria se asocia a inmunodeficiencias que pueden tener una presentación clínica fulminante, sobre todo si el desencadenante es la infección por el virus de Epstein-Barr.
- •
La ausencia de mutaciones no descarta HLH primaria, ya que las alteraciones genéticas conocidas solo justifican el 30–70% de las formas primarias.
- •
La clínica de HLH es inespecífica, incluyendo fiebre, hepatoesplenomegalia y síntomas neurológicos; los datos de laboratorio incluyen pancitopenia, elevación de la ferritina, triglicéridos y transaminasas, y disminución de la albúmina y el fibrinógeno.
- •
La edad habitual de presentación clínica de HLH es durante la lactancia.
La linfohistiocitosis hemofagocítica (HLH) fue descrita a mediados del siglo pasado por los doctores Farquhar y Claireaux utilizando el término de reticulosis hemofagocítica familiar, atribuyendo su patogenia a un defecto primario del sistema retículo endotelial1. En el momento actual, alteraciones en la función de los linfocitos T citotóxicos (CTL) y de las células natural killer (NK) se encuentran en el origen de la HLH. Además de su agrupación familiar, hoy sabemos que también existen formas esporádicas2. La HLH es un síndrome de hiperinflamación sistémica originado por una ineficaz respuesta inmunitaria ante un antígeno expuesto por las células presentadoras de antígeno (APC) que hace reverberante y amplifica la respuesta inmunitaria. Se trata de un circuito de retroalimentación positiva iniciado por una estimulación continua de CTL y células NK que provoca una producción exagerada de citocinas y una activación de los macrófagos que infiltran los tejidos destruyendo órganos vitales.
ClasificaciónLa HLH se clasifica en HLH primaria, HLH secundaria y síndromes de activación macrofágica (MAS)3.
Linfohistiocitosis hemofagocítica primaria (tabla 1)La HLH primaria incluye las formas familiares (FHLH). La FHLH tienen una incidencia aproximada de 0,12–1 casos por cada 100.000 niños por año, pero esta incidencia puede ser mayor en zonas con alta consanguinidad debido al patrón de herencia autosómica recesiva. Hasta el momento se conocen 5 enfermedades dentro de este grupo (FHLH-1, 2, 3, 4, 5), habiéndose descrito mutaciones en 4 genes. La primera alteración genómica descrita fue en el locus 9q21.3–22 en varias familias paquistaníes y más tarde fue definido como el subtipo FHLH-14. La base molecular todavía no está claramente establecida. Stepp et al describieron, en un grupo de pacientes con FHLH mutaciones en el gen de la perforina, PRF1 localizado en el cromosoma 10q21, que definieron con el subtipo FHLH-25. Las mutaciones en PRF1 representan el 20–50% de todos los casos FHLH. Muchas mutaciones en PRF1 se han identificado afectando a grupos étnicos específicos, por ejemplo c.1122G>A en pacientes turcos6. Además, las diferentes mutaciones en PRF1 originan significativa heterogeneidad clínica; por ejemplo, las mutaciones localizadas en 50delT PRF, mutaciones sin sentido o aquellas mutaciones que determinan una ausencia de la proteína perforina, asocian una edad más precoz de aparición de los síntomas de HLH7. Sin embargo, el polimorfismo C272T, que consiste en la sustitución del aminoácido alanina por valina en la posición 91 (A91V), se ha asociado a presentaciones atípicas (tardías) cuando se encuentra en homocigosis8. En heterocigosis, presente en el 8% de la población, este polimorfismo necesita de otras mutaciones adicionales, ya sea en otros genes UNC13D o en una ubicación diferente en el mismo gen PRF1. Posteriores a las descripciones hechas sobre el gen PRF1, se describieron las mutaciones en el gen UNC13D localizado en el cromosoma 17q25 (FHLH-3)9. El gen UNC13D codifica la proteína Munc13–4, crucial para la fusión de los gránulos en la sinapsis inmunológica y la liberación del contenido de las mismas. Las mutaciones en el gen UNC13D constituyen el 20–30% de los casos de FHLH. La presentación clínica de FHLH-3 se correlaciona con la cantidad de proteína Mun13–4 sintetizada10. La ausencia total de la proteína origina HLH; sin embargo, la presencia de pequeñas cantidades de la proteína se asocia a capacidad funcional y cuadros clínicos más larvados y presentaciones en edades tardías. Esta variabilidad genómica podría explicar las diferentes formas de presentación clínica, así como la necesidad de que intervengan diferentes estímulos adicionales, incluso en hermanos con similares defectos genéticos.
Lectura rápida
La linfohistiocitosis hemofagocítica (HLH), descrita a mediados del siglo pasado bajo el término de reticulosis hemofagocítica familiar, se origina por alteraciones en la función de los linfocitos T citotóxicos y de las células natural killer (NK).
La HLH se clasifica en HLH primaria, secundaria y síndromes de activación macrofágica. La HLH primaria incluye las formas familiares, de las que conocemos mutaciones en los genes PRF1, UNC13D, STX11 y STXBP2; y formas asociadas a inmunodeficiencias, como el síndrome de Griscelli tipo 2, el síndrome de Chediak-Higashi, el síndrome de Hermansky-Pudlak tipo 2, el síndrome linfoproliferativo ligado al cromosoma X, algunos casos de inmunodeficiencia combinada severa e hipogammaglobulinemia ligada al cromosoma X.
Los pacientes que presentan HLH en ausencia de mutaciones genéticas, sin asociación familiar y sin recurrencias, se clasifican como HLH secundarias. La forma más frecuente de HLH secundaria es la que se asocia a infecciones virales (virus de Epstein-Barr, citomegalovirus, virus herpes simple, virus de la inmunodeficiencia humana, virus de la gripe aviar), bacterias (micobacterias), hongos (candida) y parásitos (Leishmania). También puede aparecer en el contexto de neoplasias malignas (leucemias o linfomas), trastornos metabólicos y tratamientos prolongados con fármacos inmunosupresores.
Una variante de las formas secundarias la constituye el síndrome de activación macrofágica (MAS), que ocurre como complicación de algunas enfermedades autoinmunes, como la artritis idiopática juvenil, el lupus eritematoso sistémico o el síndrome de Kawasaki.
Los pacientes con formas primarias de HLH presentan defectos muy acusados en la citotoxicidad de los linfocitos T citotóxicos y de las células NK. Los pacientes con linfohistiocitosis hemofagocítica familiar tipo 2 presentan gránulos citotóxicos sin perforina y los pacientes con HLH familiar 3, 4 y 5 y los pacientes con inmunodeficiencias y albinismo presentan alteraciones en la degranulación de los gránulos de perforina por déficits de proteínas implicadas en la formación de los microtúbulos como Munc13-4, sintaxina 11, Munc18-2, Rab27a y Lyst. En ambos casos, alteración de la citotoxicidad o de la degranulación, el resultado será una alteración de la función citotóxica de los linfocitos T citotóxicos y de las células NK.
Los pacientes con formas secundarias la citotoxicidad y la degranulación no suelen estar afectados, sino que existe un desequilibrio en las poblaciones inmunes entre células efectoras (linfocitos T citotóxicos y de las células NK) y presentadoras (células presentadoras de antígeno), provocado por los patógenos infecciosos.
En todos los casos, en los síndromes hemofagocíticos hay una repuesta inflamatoria sistémica exagerada que, sin embargo, es incapaz de eliminar el desencadenante inicial y provoca una gran activación de los linfocitos T citotóxicos, macrófagos y de las células NK que infiltran órganos vitales, proliferan y liberan citocinas (interferón gamma, factor de necrosis tumoral alfa, interleucina 1 (IL-1), IL-6, IL-8, IL-10, IL-18, factor estimulante de colonias granulocíticas y microcíticas, sCD25).
Las manifestaciones clínicas son la consecuencia de la activación de las células del sistema inmunitario, la infiltración en los órganos y tejidos y de la liberación de citocinas. Incluyen fiebre prolongada rebelde a tratamiento antibiótico, hepatoesplenomegalia, adenopatías, rash cutáneo y síntomas neurológicos, como irritabilidad o convulsiones.
Los hallazgos de laboratorio incluyen citopenias, hiperferritinemia, elevación de transaminasas, hipofibrinogenemia, hipertrigliceridemia, hipoalbuminemia, hiponatremia, pleocitosis e hiperproteinorraquia.
Los hallazgos de inmunológicos incluyen elevación del receptor soluble de la IL-2 y la disminución de la actividad citotóxica de las células NK.
La hemofagocitosis suele estar ausente al inicio de los síntomas clínicos, por lo que resulta un hallazgo prescindible para el diagnóstico de HLH.
A pesar de que la presentación clínica habitual ocurre en niños pequeños, cada vez se describen más casos en pacientes de más edad, incluso en adultos. Hasta en el 14% de los adultos que presentan HLH, se han descrito mutaciones en los genes PRF1, MUNC13-4 y STXBP2, dando lugar a defectos de citotoxicidad menos severos que causan formas atípicas de presentación de HLH más allá de la edad pediátrica.
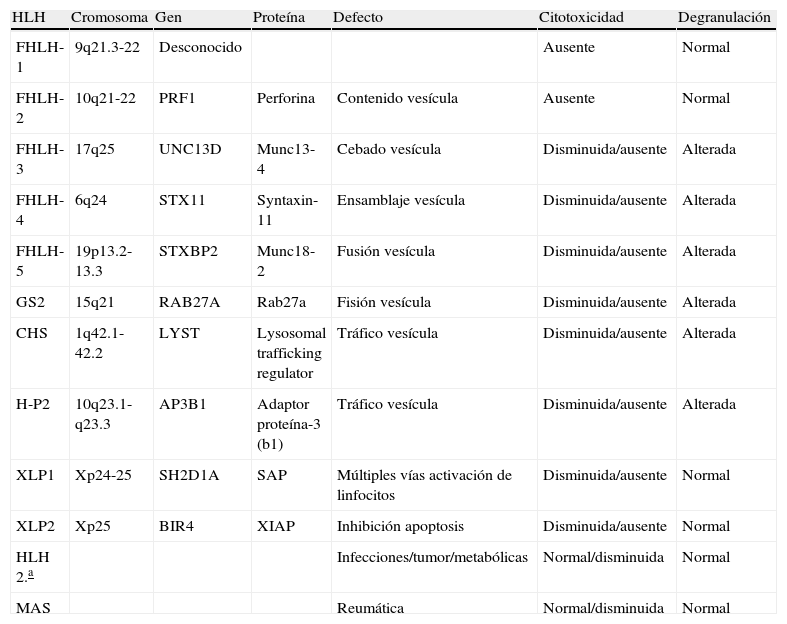
Alteraciones genéticas y defectos fisiopatológicos en la linfohistiocitosis hemofagocítica.
| HLH | Cromosoma | Gen | Proteína | Defecto | Citotoxicidad | Degranulación |
| FHLH-1 | 9q21.3-22 | Desconocido | Ausente | Normal | ||
| FHLH-2 | 10q21-22 | PRF1 | Perforina | Contenido vesícula | Ausente | Normal |
| FHLH-3 | 17q25 | UNC13D | Munc13-4 | Cebado vesícula | Disminuida/ausente | Alterada |
| FHLH-4 | 6q24 | STX11 | Syntaxin-11 | Ensamblaje vesícula | Disminuida/ausente | Alterada |
| FHLH-5 | 19p13.2-13.3 | STXBP2 | Munc18-2 | Fusión vesícula | Disminuida/ausente | Alterada |
| GS2 | 15q21 | RAB27A | Rab27a | Fisión vesícula | Disminuida/ausente | Alterada |
| CHS | 1q42.1-42.2 | LYST | Lysosomal trafficking regulator | Tráfico vesícula | Disminuida/ausente | Alterada |
| H-P2 | 10q23.1-q23.3 | AP3B1 | Adaptor proteína-3 (b1) | Tráfico vesícula | Disminuida/ausente | Alterada |
| XLP1 | Xp24-25 | SH2D1A | SAP | Múltiples vías activación de linfocitos | Disminuida/ausente | Normal |
| XLP2 | Xp25 | BIR4 | XIAP | Inhibición apoptosis | Disminuida/ausente | Normal |
| HLH 2.a | Infecciones/tumor/metabólicas | Normal/disminuida | Normal | |||
| MAS | Reumática | Normal/disminuida | Normal |
CGS: síndrome de Chediak Higashi; FHLH-1: linfohistiocitosis hemofagocítica familiar tipo 1; FHLH-2: linfohistiocitosis hemofagocítica familiar tipo 2; FHLH-3: linfohistiocitosis hemofagocítica familiar tipo 3; FHLH-4: linfohistiocitosis hemofagocítica familiar tipo 4; FHLH-5: linfohistiocitosis hemofagocítica familiar tipo 5; GS2: síndrome de Griscelli tipo II; HLH 2.a: linfohistiocitosis hemofagocítica secundaria; H-P2: síndrome de Hermansky-Pudlak II; MAS: síndrome de activación macrofágica; XLP1: síndrome linfoproliferativo ligado a X tipo 1; XLP2: síndrome linfoproliferativo ligado a X tipo 2.
La FHLH-4 afecta a mutaciones en el gen STX11. Inicialmente fue descrito en varias familias kurdas11. El gen STX11 se encuentra localizado en el cromosoma 6q24 y codifica la proteína Stx11. El déficit de esta proteína determina una degranulación deficiente de CTL y NK, que se restaura parcialmente tras estimulación con interleucina 2 (IL-2). Esta situación origina una presentación clínica más tardía y leve que FHLH-2 y FHLH-312. La expresión de CD107a, tal y como describieron Bryceson et al, también ayuda al diagnóstico de los déficit de degranulación mediados por sintaxina-1113.
Además de las formas familiares, la HLH primaria se asocia a algunas inmunodeficiencias como el síndrome de Griscelli tipo 2 (GS2)14, el síndrome de Chediak-Higashi (CHS)15, el síndrome de Hermansky-Pudlak tipo 2 (H-P2)16, el síndrome linfoproliferativo ligado al cromosoma X (XLP)17, algunos casos de inmunodeficiencia combinada severa18 e hipogammaglobulinemia ligada al cromosoma X19. Varias de estas inmunodeficiencias, como los GS2, CHS y H-P2, están asociados a anormalidades en las proteínas implicadas en el transporte de los gránulos citolíticos (tal como los FHLH), así como en los melanosomas, lo que origina un cuadro clínico de HLH que incluye diferentes grados de albinismo20. El GS2 se caracteriza por mutaciones en el gen RAB27A localizado en el cromosoma 15q21. Estas mutaciones condicionan que los gránulos citotóxicos, una vez polarizados en los microtúbulos, no puedan desplazarse hacia la sinapsis inmunológica. En modelos murinos el déficit de RAB27A solo origina HLH en asociación a un desencadenante infeccioso, viral. Este dato podría explicar la heterogeneidad de la presentación clínica.
El CHS es causado por mutaciones en el gen transportador de lisosoma LYST localizado en el cromosoma 1q42.1–42.2. Esta proteína es fundamental para los procesos de fusión y fisión de los lisosomas; el resultado final es la ausencia de secreción del contenido de los lisosomas en la sinapsis inmunológica21. Los pacientes con CHS presentan albinismo, infecciones bacterianas recurrentes y episodios de HLH. Se caracterizan por presentar grandes lisosomas en todos los tipos celulares. El síndrome H-P2 origina albinismo parcial y HLH. El defecto se encuentra en el gen adaptador de proteína-3, AP3B1, que codifica para la subunidad beta del complejo AP-3. La ausencia de AP-3 afecta al transporte de las vesículas del citoplasma a la membrana en ciertos tipos de células, incluyendo melanosomas, plaquetas, CTL y las células NK22.
Los pacientes con XLP tienen una especial sensibilidad a las infecciones por virus de Epstein-Barr (VEB), lo que resulta en una respuesta inmunitaria exagerada y a menudo fatal, en forma de HLH fulminante23. En ausencia de tratamiento, los pacientes fallecen entre los 5–20 años de edad. El XLP se ha relacionado con mutaciones en el gen SH2D1A, que codifica la proteína SAP (proteína asociada a estimulación), implicada en la señalización de la activación linfocítica24. SAP tiene un papel importante en el desarrollo, la diferenciación y la función efectora de los linfocitos B, CTL, CD4 y NK. Otra alteración genética que asocia XLP se ha observado en el gen BIRC4. Este gen codifica una proteína implicada en la apoptosis celular.
Las alteraciones genéticas conocidas solo justifican el 30–70% de las HLH primarias, luego la ausencia de mutaciones no descarta en su totalidad el diagnóstico de HLH primaria, y hace complejo tanto el diagnóstico como el consejo genético25.
Linfohistiocitosis hemofagocítica secundariaLos pacientes que presentan HLH en ausencia de alteraciones y/o indicadores de predisposición genética, tales como asociación familiar o episodios recurrentes, se clasifican como HLH secundaria. A pesar de que los datos son escasos, se estima que su frecuencia es mayor que las formas primarias. Sin embargo, para el diagnóstico hay que tener un elevado grad[o de sospecha, ya que puede pasar inadvertido y solaparse con otras entidades clínicas, como la sepsis y el fallo multiorgánico. La forma más frecuente de HLH secundaria es la que se asocia a infecciones26. Los agentes infecciosos implicados son virus, como el VEB, el citomegalovirus, el virus herpes simple, el virus de la inmunodeficiencia humana, el virus de la gripe aviar; bacterias como micobacterias, micoplasmas; parásitos como Leishmania y Plasmodium, y hongos como candida y criptococo. La asociación de HLH e infección es importante, ya que los pacientes con sepsis grave con signos clínicos de HLH podrían beneficiarse del tratamiento para esta entidad27.
A pesar de que el VEB y la Leishmania son los agentes infecciosos que más frecuentemente originan HLH secundaria, no es infrecuente que los episodios agudos en pacientes con HLH primaria también estén provocados por estas infecciones. También neoplasias malignas, como leucemias o linfomas, en particular linfomas de células T y algunos tumores sólidos, pueden provocar excepcionalmente HLH. Trastornos metabólicos, incluyendo déficits múltiples de sulfatasas, lisinuria, trastornos del metabolismo del ácido propiónico y enfermedad de Wolman, pueden asociar HLH. Por último, el tratamiento inmunosupresor, habitual en el tratamiento de algunos cánceres, enfermedades autoinmunes y en el trasplante de órgano sólido y hematopoyético, puede predisponer a desarrollar HLH secundaria. Los mecanismos que subyacen a estas asociaciones no están bien establecidos28.
Síndrome de activación macrofágicaEl síndrome de activación macrofágica (MAS) es una complicación grave de enfermedades autoinmunes e inflamatorias, que puede considerarse como una variante de HLH secundaria. Habitualmente, el curso clínico del MAS es indistinguible de la HLH29. Se caracteriza por fiebre, hepatoesplenomegalia, linfadenopatías, hiperferritinemia, disfunción hepática y coagulopatía. La disfunción cardíaca y neurológica puede aparecer durante el cuadro clínico. Además, recientemente se ha demostrado cómo los pacientes con MAS presentan alteraciones cuantitativas y funcionales de las células NK, al igual que otros HLH. Sin embargo, los pacientes con MAS suelen mostrar neutrofilia o trombocitosis en etapas iniciales de la enfermedad, hallazgos inhabituales en HLH primaria. La incidencia de MAS oscila del 7 al 30% en pacientes con artritis idiopática juvenil sistémica activa (enfermedad de Still), con una mortalidad de hasta el 22%, según las series30. Hasta en un 7% puede ser la forma inicial de presentación. Otras enfermedades reumáticas, como la enfermedad de Kawasaki y el lupus eritematoso sistémico, pueden presentar MAS en algún momento de su evolución. Los virus, los fármacos como los antiinflamatorios no esteroideos, el metotrexato y las sales de oro, incluso inhibidores de factor de necrosis tumoral alfa (TNF-α) pueden actuar como factores desencadenantes de MAS.
FisiopatologíaFisiología de la activación y expansión del sistema inmunitarioEl sistema inmunitario innato constituye la primera línea de defensa frente a las infecciones y los tumores. Este papel está mediado fundamentalmente a través células dendríticas, macrófagos y células NK. El papel de las células NK ha sido descrito recientemente y adquiere una trascendencia vital en la patogenia de la HLH31. Las células NK se definen como un linfocito del sistema inmunitario innato, que expresan el marcador de superficie CD56, cuya función está regulada a través del equilibrio dinámico entre diferentes receptores que reciben estímulos activatorios e inhibitorios. Se diferencian 2 poblaciones de células NK: a) las células NK dim, mayoritarias caracterizadas por la expresión de CD16 (receptor para la fracción Fc de las inmunoglobulinas), con una función fundamentalmente citotóxica, y b) las células NK bright, minoritarias, que carecen del receptor CD16, y que se caracterizan funcionalmente por la capacidad de liberar grandes cantidades de citocinas, fundamentalmente interferón gamma (IFN-γ), TNF-α y factor estimulante de colonias granulocíticas y monocíticas (GM-CSF). Estos, a su vez, modulan al sistema inmunitario innato y adaptativo, dirigiendo la respuesta inmunitaria hacia un fenotipo activatorio, Th14. Las células NK bright poseen receptores de quimiocinas que permite que sean atraídas hacia órganos linfoides secundarios. Las células NK pueden ser activadas directamente a través del contacto célula-célula, o indirectamente a través de la secreción de citocinas como IL-12, IL-15 e IL-18.
Por otro lado, los linfocitos B y T constituyen las principales células del sistema inmunitario adaptativo, y son responsables de la eliminación de los patógenos que no han sido eliminados por el sistema inmunitario innato, así como por el desarrollo de memoria inmunológica. Cuando una APC activa a través de este reconocimiento a los linfocitos T (CD4 y CD8) con especificidad para el antígeno apropiado, estos proliferan y se diferencian para proceder a la rápida eliminación del antígeno. La activación de las células T origina la producción de citocinas proinflamatorias que, a su vez, atraen a otras células del sistema inmunitario (linfocitos B y T, y macrófagos), activando el sistema del complemento, la opsonización, la fagocitosis y la lisis de los antígenos por las células del sistema inmunitario innato. Ambos sistemas, el adaptativo y el innato, colaboran estrechamente para eliminar a los patógenos. El fin de esta respuesta expansiva está determinado por un proceso de contracción de la respuesta inmunitaria donde el 95% de los linfocitos estimulados específicos de antígenos entran en apoptosis y desaparecen, permaneciendo una pequeña población de linfocitos con propiedades memoria (fig. 1).

Modelo de expansión y contracción de la respuesta inmunitaria ante un estímulo antigénico. La respuesta normal se caracteriza por una expansión controlada seguida de una rápida contracción. Sin embargo, en HLH la expansión es incontrolada y se caracteriza por la ausencia de contracción, por lo que se perpetúa el estado inflamatorio. HLH: llinfohistiocitosis hemofagocítica.
La actividad citotóxica de los CTL y las células NK está controlada por 2 mecanismos conocidos hasta el momento: la liberación de gránulos que contienen proteínas líticas, como la perforina y las granzimas, en la sinapsis inmunológica y la apoptosis inducida por receptor32. En el proceso de secreción de los gránulos con lisosomas líticos son fundamentales la organización de los microtúbulos y la formación, la polarización, la vehiculización y la fusión de los gránulos. En este proceso son fundamentales proteínas como Lyst, Rab 27, AP3B1, Stx11, y Munc13–4. La degranulación se puede detectar por la aparición de la proteína de membrana asociada a lisosoma, CD107a, en la membrana celular. La perforina se almacena en los lisosomas secretores de las células citotóxicas NK, CTL, también en las células T reguladoras y en las células NKT16. Estos gránulos contienen granzimas, serglicano y careticulina, y enzimas y moléculas asociadas a la membrana de los lisosomas (CD63, CD107a y CD107b). La presencia de perforina es esencial para inducir apoptosis en la célula diana a través de la activación de la cascada de caspasas. Las granzimas son serinaproteasas con el potencial de desencadenar la muerte celular en las células tumorales y en células infectadas por virus33.
El reconocimiento entre la células efectora (CTL y/o NK) y la célula diana (APC, célula infectada o tumoral) es dependiente del contacto y se realiza en la sinapsis inmunológica. Tras el reconocimiento, las células efectoras liberan los gránulos citotóxicos que contienen perforinas y granzimas al espacio intercelular, donde ejercen su acción citotóxica. Este fenómeno se denomina gráficamente «el beso de la muerte». Esta actividad citotóxica no es exclusiva sobre la célula infectada o transformada, sino que también es efectiva frente a las células presentadoras de antígeno (APC), de manera que proporciona un importante mecanismo de retroalimentación negativa para limitar la respuesta inmune mediada por los linfocitos T y las células NK. En ausencia de este mecanismo de retroalimentación, las APC continúan estimulando los CTL y las NK, lo que origina una hiperproducción de citocinas, como IFN-γ y TNF-α, que tienen un papel clave en la activación de los macrófagos.
Respuesta inmunitaria y citotóxica en linfohistiocitosis hemofagocíticaAtendiendo a la fisiología de la respuesta inmunitaria, podemos definir la HLH como una afección donde hay 2 defectos asociados: a) una hiperexpansión de la respuesta inmunitaria por una estimulación antigénica continua, y b) un defecto en la fase de contracción de la misma por persistencia del estímulo antigénico (fig. 1).
La HLH se describe como una repuesta inflamatoria sistémica ineficaz caracterizada por la participación de células hiperactivadas (CTL, macrófagos y NK) que infiltran diferentes órganos y proliferan en ellos secretando una gran cantidad de citocinas. Estos factores son los responsables de las manifestaciones clínicas34. Los CTL, las NK y los macrófagos activados infiltran el hígado, la médula ósea, el bazo, el sistema nervioso central secretando citocinas (IL-1, IL-6, IFN-γ y TNF-α) y mostrando una exagerada actividad fagocítica. El IFN-γ y el TNF-α tienen efectos tóxicos sobre las células hematopoyéticas, originando citopenia. El TNF-α inhibe la lipoproteína lipasa, causando hipertrigliceridemia, y las citocinas IL-1, IL-6 y TNF-α son responsables de la aparición de fiebre. Los macrófagos activados secretan ferritina, así como el activador del plasminógeno. Las células NK y los CTL activados liberan el exceso del receptor para la IL-2 al plasma, siendo un factor determinante para el diagnóstico en el laboratorio de actividad de HLH. Recientes datos obtenidos de la experimentación en animales indican que el IFN-γ es la citocina clave implicada en esta cascada inflamatoria y que su bloqueo podría ser un tratamiento efectivo para los pacientes con HLH35–40. Otras citocinas que desempeñan un papel relevante son TNF-α, IL-1, IL-6, IL-8, IL-10, IL-18, factor estimulante de colonias granulocíticas y monocíticas, sCD25. La hipersecreción de citocinas generada por la activación incontrolada de CTL, NK y macrófagos parece ser la base de la mayor parte de la disfunción orgánica progresiva que conduce a la muerte en los pacientes gravemente afectados37. Quimiocinas tales como la proteína quimiotáctica de monocitos y la proteína inflamatoria de macrófagos son incluso más precoces que la elevación de ferritina, lactatodeshidrogenasa o transaminasas. La infiltración de CTL, NK, APC y macrófagos origina hepatoesplenomegalia, adenopatías, infiltración pulmonar, neurológica y hemofagocitosis en médula ósea.
De acuerdo con este modelo, la disfunción de las células NK y CTL conduce a una prolongación de la respuesta inmunitaria, con una «tormenta» de citocinas y al fenotipo HLH. Este modelo fue propuesto por Badovinac et al en modelos murinos sin perforina (PRF–/–) utilizando como estímulo infecciones mediante el virus de la coriomeningitis linfocítica o la bacteria Lysteria monocytogenes38.
Los pacientes con FHLH y los pacientes con inmunodeficiencias que asocian albinismo (CHS, GS2 y el H-P2) presentan defectos en la citotoxicidad de CTL y células NK. Los pacientes con FHLH-2 presentan gránulos citotóxicos vacíos, no contienen las proteínas perforina y granzimas, y los pacientes con FHLH-3, 4 y 5 y los pacientes con inmunodeficiencias que asocian albinismo presentan alteraciones en la biogénesis, polarización, el transporte o la exocitosis de los gránulos citotóxicos (fig. 2). A pesar de que la fisiopatología es diferente el resultado funcional es el mismo, ausencia de actividad citotóxica frente a las células dianas y, por tanto, persistencia de los estímulos inflamatorios, mediante la hipersecreción de citocinas.
. Patogénesis de la FHLH (enfermedad/ gen y defecto): FHLH-2 mutaciones en PRF1 que origina una ausencia de las proteínas perforina y granzima en las vesículas; FHLH-3, 4 y 5, GS2, CHS, H-P2 y mutaciones en genes que regulan el trasporte de las vesículas del citoplasma a la sinapsis inmunológica.")
Sinapsis inmunológica formada por la célula efectora, NK o CTL y la célula diana (célula infectada, célula tumoral, célula presentadora de antígeno). Patogénesis de la FHLH (enfermedad/ gen y defecto): FHLH-2 mutaciones en PRF1 que origina una ausencia de las proteínas perforina y granzima en las vesículas; FHLH-3, 4 y 5, GS2, CHS, H-P2 y mutaciones en genes que regulan el trasporte de las vesículas del citoplasma a la sinapsis inmunológica.
Además de estos mecanismos, se postulan como mecanismos relacionados con el desarrollo de HLH los mecanismos implicados en la liberación de IL-10, TGF-α y FAS ligando, el receptor NKG2D en las células NK y su incapacidad para suprimir las células T39,40.
Por el contrario, la fisiopatología de HLH en las inmunodeficiencias por déficit de las proteínas SAP y XIAP está desencadenada casi exclusivamente por la infección por VEB. Por ejemplo, el déficit de la proteína SAP en los CTL hace que la interacción con los linfocitos B, dianas del VEB, sea insuficiente y no pueda controlarse la infección, originando el síndrome linfoproliferativo y apareciendo HLH como manifestación del mismo.41-44
La fisiopatología de las HLH secundarios es menos conocida. En la mayoría de los casos, la degranulación de los linfocitos y la citotoxicidad no están afectados. Sin embargo, sí está afectado el equilibrio entre los CTL/NK y las APC, predominando la activación de estas últimas por patógenos intracelulares a través de los receptores tipo Toll (TLR). En este sentido, se ha descrito en modelos animales el papel protector de la citocina IL-10 al bloquear la estimulación inducida por el receptor TLR-941.
ClínicaEl cuadro clínico de HLH es bastante característico, pero la presentación inicial no es específica. Las primeras manifestaciones clínicas son similares a las infecciones comunes, neoplasias, fiebre de origen desconocido, enfermedades autoinmunes y trastornos inflamatorios. Posteriormente, aparecen signos típicos de HLH, como fiebre prolongada que no responde a tratamiento antibiótico, hepatoesplenomegalia y síntomas neurológicos (irritabilidad, disminución del nivel de consciencia, convulsiones, ataxia), presentes hasta en el 30% de los pacientes. Otras manifestaciones clínicas menos frecuentes son las adenopatías, el rash, los edemas y la ictericia.
Los hallazgos de laboratorio incluyen citopenias, generalmente comienza con trombocitopenia que evoluciona a pancitopenia, hiperferritinemia, elevación de transaminasas, hipofibrinogenemia, hipertrigliceridemia, hipoalbuminemia e hiponatremia. Muchos pacientes con HLH muestran signos de coagulación intravascular diseminada. Los hallazgos inmunológicos incluyen elevación del receptor soluble de la IL-2 y la disminución de la actividad citotóxica de las células NK45. La hemofagocitosis suele estar ausente al inicio de la enfermedad, apareciendo posteriormente. Sin embargo, tenemos que tener en cuenta que el diagnóstico de HLH no depende en absoluto de este hallazgo.
A pesar de que la edad habitual de presentación de HLH es menor de 2 años, cada vez se describen más casos de HLH en niños mayores, incluso en adultos, con manifestaciones clínicas tan variadas como anemias aplásicas, síndrome linfoproliferativo autoinmune, hipogammaglobulinemia, granulomatosis pulmonar, cerebral y hepática o encefalitis aséptica47. Hasta en el 14% de los adultos que presentan HLH se observan mutaciones en los genes PRF1, MUNC13-4 y STXBP2, con actividad parcial de las proteínas (mutaciones hipomorfas) dando lugar a defectos de citotoxicidad menos severos que causan formas atípicas de presentación de HLH más allá de la edad pediátrica.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.