








Puntos clave
- •
El síndrome nefrítico se define por hematuria, proteinuria, oliguria y edemas con grado variable de hipertensión arterial e insuficiencia renal.
- •
La causa más frecuente, que no única, es la glomerulonefritis aguda postestreptocócica.
- •
Los síntomas son disminución de la diuresis, orinas oscuras y ganancia de peso; en la exploración física, hipertensión arterial y edemas.
- •
Se caracteriza por elevación de la urea más que de creatinina con hematuria glomerular y alteraciones inmunológicas, siendo la más característica el descenso del C3. La biopsia renal da el diagnóstico etiológico, aunque en los niños no suele ser necesaria.
- •
El tratamiento depende de la causa etiológica. En la mayoría de las ocasiones, es de soporte: restricción de líquidos y sodio, diuréticos y antihipertensivos. Si no es suficiente el manejo conservador, puede precisar técnica dialítica. El tratamiento con eculizumab es una opción en la glomerulopatía C3.
- •
El pronóstico depende de la etiología, siendo en general benigno.
Lectura rápida
La glomerulonefritis postestreptocócica (GNAPE) es la causa más frecuente de síndrome nefrítico en el mundo. Tiene una mayor incidencia en países en vías de desarrollo. El riesgo de la GNAPE es mayor en los niños entre 5 y 12 años y adultos mayores de 60 años.
La GNAPE se produce por inmunocomplejos inducidos por cepas nefritogénicas del estreptococo betahemolítico del grupo A (EGA). Los 2 antígenos nefritogénicos son el receptor de la plasmina asociado a nefritis y la exotoxina B pirogénica del estreptococo.
Aunque la biopsia no suele ser necesaria, la característica anatomopatológica de la GNAPE son los depósitos subepiteliales con forma de joroba en la microscopia electrónica. En la inmunofluorescencia, encontramos depósitos de IgG y C3, con un patrón granular difuso en el mesangio y en las paredes capilares del glomérulo, y en la microscopia óptica se encuentra infiltrados celulares y proliferación endocapilar difusa.
Aunque la mayoría de los hallazgos clínicos incluyen edema, hematuria macroscópica e hipertensión, la presentación de la GNAPE puede variar de asintomática (muy frecuente aunque difícil de diagnosticar), a síndrome nefrítico florido (hematuria macroscópica, proteinuria, edema, hipertensión arterial y fallo renal agudo). El diagnóstico se puede sospechar porque el paciente está hipertenso, con edema periférico y porque la hematuria precede al fallo renal.
Los hallazgos de laboratorio incluyen análisis de orina anormal (hematíes dismórficos, grado variable de proteinuria, cilindros hemáticos y piuria), serología positiva para anticuerpos frente a los antígenos del estreptococo e hipocomplementemia (descenso de C3).
GNAPE se diagnostica ante un síndrome nefrítico demostrando el antecedente de una infección reciente por EGA.
El diagnóstico diferencial se realiza con glomerulonefritis, que tienen un comienzo similar, con algún matiz que obliga a realizar la biopsia renal. La histología renal nos dará el diagnóstico de la lesión glomerular. La glomerulonefritis membranoproliferativa (GNMP) es la anatomía patológica que nos encontramos con más frecuencia.
Clásicamente, se definían 3 tipos de acuerdo con la localización de los depósitos, siendo el tipo II el de peor pronóstico. Recientemente, se han pasado a diferenciar de acuerdo a la presencia exclusiva de C3 lo que indica activación del complemento por la vía alternativa y peor pronóstico (glomerulopatía C3) o depósitos múltiples. La glomerulonefritis mesangial IgA, púrpura de Schönlein-Henoch, el lupus y la enfermedad de Wegener también pueden comenzar como una glomerulonefritis rápidamente progresiva.
No hay una terapia específica para tratar la GNAPE. El manejo es de soporte y se centra en tratar la sobrecarga de volumen. Las medidas terapéuticas incluyen restricción de líquidos y de sal, y tratamiento con diuréticos y erradicación del EGA. En las GNMP tipo II que se corresponden con glomerulopatía C3, con evolución rápidamente progresiva, los anticuerpos monoclonales y, más concretamente, el eculizumab son una opción terapéutica prometedora.
En pacientes con fallo renal agudo, puede ser necesaria la diálisis. Las indicaciones de la diálisis son:
- –
Rápido deterioro de la función renal con riesgo de complicaciones secundarias a uremia, en general, cifras superiores a 200mg/dl.
- –
Edema agudo de pulmón con oliguria sin respuesta a diuréticos.
- –
Trastornos hidroelectrolíticos con riesgo vital refractarios a medidas conservadores.
En pacientes con hipertensión arterial, se pueden administrar diuréticos para provocar una diuresis precoz y una reducción en la tensión arterial. En raras ocasiones, el paciente puede presentar encefalopatía hipertensiva y es necesario un tratamiento de emergencia para reducir lentamente la presión arterial.
La mayoría de los pacientes, especialmente los niños, se recuperan totalmente y la resolución comienza en unos 15 días. Las cifras de complemento se normalizan en 6-8 semanas. Un pequeño número de pacientes tienen complicaciones tardías (hipertensión, proteinuria e insuficiencia renal).
El síndrome nefrítico (SN) es una constelación de manifestaciones clínicas causadas por un proceso inflamatorio en el glomérulo. Produce disminución de la filtración glomerular, con retención de sodio y agua. Se acompaña de hipertensión arterial (HTA) e insuficiencia renal en grado variable, con oliguria y edema. Se caracteriza por hematuria glomerular con un sedimento activo con hematíes dismórficos, cilindros hemáticos y granulosos. La proteinuria está presente en grado variable. La hematuria puede ser esporádica, intermitente o persistente; micro o macroscópica. El SN puede ocurrir como un proceso renal aislado o como parte de una enfermedad sistémica o hereditaria. En la edad pediátrica, la causa más frecuente es la glomerulonefritis aguda postestreptocócica, aunque no debemos olvidar que existen otras causas, en general de peor pronóstico, en la cuales será muy importante realizar un diagnóstico precoz para plantear una opción terapéutica1.
AnamnesisEl comienzo de un SN agudo puede ser brusco (el paciente refiere fiebre, cefalea y dolor abdominal) o progresivo con edema periférico, ganancia de peso y astenia. El síntoma prínceps es la hematuria, que puede acompañarse o no de oliguria. La orina tiene espuma como resultado de la eliminación de proteínas que tienen una acción reductora de la tensión superficial de la orina. Las preguntas irán dirigidas a diferenciar la hematuria glomerular de la urológica o urotelial. La hematuria glomerular se describe como marrón, té o color coca-cola, mientras que la urológica se describe como roja con coágulos. Rara vez refieren dolor leve en flanco, espalda o abdomen, al igual que en la hematuria urológica, donde es la norma. La pregunta sobre antecedente cercano de fiebre, infección del tracto respiratorio superior o de la piel es obligada, y si la respuesta es positiva nos orienta a una glomerulonefritis aguda postestreptocócica o postinfecciosa. En este punto, es importante buscar signos de alarma que orientan a otras etiologías: edad menor de 4 años o superior a 15, historia familiar de enfermedades glomerulares, historia previa de síntomas similares, evidencia de enfermedad extrarrenal o sistémica y/o evidencia de enfermedad renal crónica con HTA2-5.
Examen físicoLos hallazgos más característicos son el edema periférico y la HTA. El edema y la HTA sistémica son secundarios a la expansión del volumen. El edema es, en general, moderado, pero el paciente puede llegar a presentar anasarca, insuficiencia cardiaca con edema agudo de pulmón y edema cerebral. La HTA suele ser moderada pero en ocasiones puede presentarse como HTA maligna con daño en órganos diana. Otros hallazgos que nos van a orientar a una etiología sistémica o hereditaria del SN son: en la piel, presencia de rash típico en ala de mariposa (lupus eritematoso sistémico), lesiones elevadas palpables purpúricas (vasculitis, entre las que se encuentra la púrpura de Schönlein-Henoch y angioqueratomas presentes en la enfermedad de Fabry), y en articulaciones, artritis, hiperelasticidad, rigidez, que pueden indicar la presencia de una enfermedad reumática, colagenosis o vasculitis. El resto de la exploración física es inespecífica.
Fisiopatología del daño glomerular en el síndrome nefrítico en las diferentes etiologíasEl SN es secundario a la inflamación del glomérulo. El daño glomerular puede ser el resultado de alteraciones genéticas, inmunológicas, en la perfusión o en la coagulación y sistema de complemento. Los trastornos genéticos del glomérulo resultan de las mutaciones en el ADN del exón que codifica las proteínas localizadas en el glomérulo, en el intersticio y en el epitelio tubular. Las alteraciones inmunológicas están mediadas tanto por mecanismos humorales como celulares. Los mecanismos mediados por anticuerpos son de 2 tipos: anticuerpos contra los componentes de la estructura del glomérulo (p. ej., en la enfermedad de Wegener contra la membrana basal) o complejos antígenoanticuerpo que se escapan al sistema retículo endotelial y que, a su vez, se depositan en el glomérulo (nefropatía IgA), o bien mediante la interacción antígeno-anticuerpo in situ. Este último mecanismo puede producir o no la liberación de inmunocomplejos circulantes. Otros mecanismos del daño glomerular incluyen el sistema del complemento y la coagulación, la apoptosis y la síntesis alterada de citocinas, que conllevan la entrada de los leucocitos circulantes. El sistema del complemento se puede activar por la vía clásica, vía lecitina o por la vía alternativa. La ruta de activación puede guiar al clínico hacia el diagnóstico subyacente. Una activación por la vía alternativa produce una disminución de los niveles de C3 sérico con niveles de C4 normales, hecho que se ve en la glomerulonefritis aguda postestreptocócica6-8. Una vez que han comenzado los sucesos, los mecanismos secundarios de daño glomerular comienzan con una cascada de mediadores inflamatorios que son los responsables del incremento de la permeabilidad a las proteínas y la disminución del filtrado glomerular, y eso provoca las alteraciones estructurales del glomérulo, con hipercelularidad, trombosis, necrosis y formación de semilunas. Se produce un aumento de la reabsorción de sal en la nefrona distal, especialmente en el túbulo cortical y eso conlleva una retención de líquidos y sal, con un sistema renina-angiotensina-aldosterona que funciona normalmente (fig. 1)2,5,9.
Pruebas de laboratorioEstudios urinarios

La hematuria es fácilmente reconocible con un resultado positivo para sangre en la tira de orina o por la visión directa. La hematuria glomerular se acompaña con frecuencia de proteinuria. El rango de la proteinuria puede variar de bajo grado (< 500mg/día) a proteinuria nefrótica (> 3.000mg/día). La proteinuria se debe cuantificar en una orina de 24h. Otras alternativas son los índices proteinuria/ creatinina en orina de una micción, que da una idea bastante aproximada de la magnitud del problema. El examen microscópico de la orina en el síndrome nefrótico revela un número variable de hematíes libres. Generalmente, existe un botón hemático en el fondo del tubo centrifugado de orina. Los cilindros hemáticos son un hallazgo definitivo de afectación glomerular, aunque no se visualizan en todas las ocasiones. Pueden aparecer también cilindros granulosos e hialinos, sobre todo si la proteinuria es elevada. Una característica identificativa del sangrado glomerular son los hematíes dismórficos con protrusiones, burbujas y vesículas (fig. 2).
Estudios hemáticos
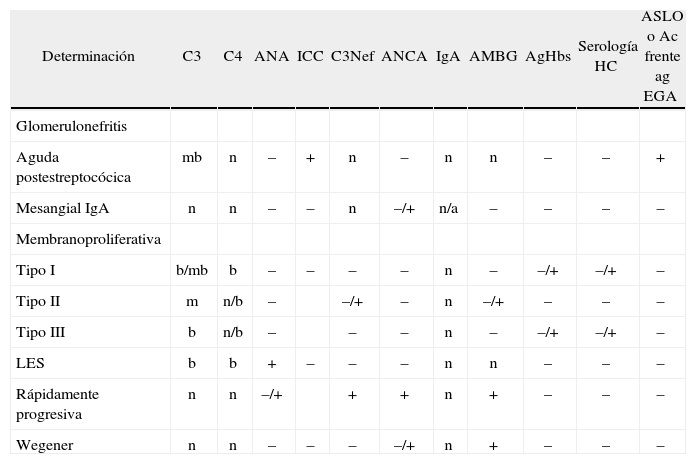
Los estudios rutinarios de laboratorio incluyen recuento sanguíneo completo, electrolitos, urea, creatinina y perfil hepático. La velocidad de sedimentación globular y la proteína C reactiva están elevadas. Hay que realizar un filtrado glomerular. Generalmente, se realiza con una recogida de orina de 24h para hacer aclaramiento de creatinina correcta, ya que el filtrado glomerular estimado por la fórmula de Schwartz modificada basa el aclaramiento en una función renal estable, con cifras de creatinina sin variaciones rápidas, y nos puede dar datos erróneos. Ante la sospecha de un SN, es obligado un estudio inmunológico que incluirá complemento y anticuerpos antinucleares (ANA). El C3, componente de vía clásica y alternativa y C4, componente de la vía clásica solo nos servirán para enfocar el trastorno; de esta forma, definiremos el SN con niveles normales de complemento y con niveles alterados (fig. 3). Niveles bajos de C3 con niveles normales de C4 indican glomerulonefritis aguda postestreptocócica o postinfecciosa, o glomerulonefritis membranoproliferativa (GNMP), mientras que bajos niveles de ambos indican glomerulonefritis postinfecciosa, lupus eritematoso sistémico, GNMP asociada a hepatitis C tipo I o crioglobulinemia mixta. La presencia de ANA positivos nos orienta hacia una enfermedad sistémica; en este caso, otros estudios inmunológicos a incluir serán: anti-ADN, anti-Sm y anti-Ro para el diagnóstico de enfermedades de colágeno, fundamentalmente el lupus eritematoso sistémico; anticuerpos perinucleares anticitoplasma de neutrófilos y anticuerpos citoplasmáticos anticitoplasma de neutrófilo para el diagnóstico de vasculitis; anticuerpos antimembrana basal glomerular para descartar la enfermedad de Wegener o el síndrome de Googpasture (tabla 1). El SN con C3 bajo puede ser secundario a un número de enfermedades infecciosas; incluiremos, según la política de vacunas de la comunidad, antecedentes personales y edad, serologías de hepatitis virales B y C, sífilis y virus de la inmunodeficiencia humana. Otras enfermedades infecciosas que se deben considerar son endocarditis, infecciones bacterianas persistentes, como abscesos, o infecciones de shunt vasculares.

Diagnóstico diferencial del síndrome nefrítico: alteraciones inmunológicas.
| Determinación | C3 | C4 | ANA | ICC | C3Nef | ANCA | IgA | AMBG | AgHbs | Serología HC | ASLO o Ac frente ag EGA |
| Glomerulonefritis | |||||||||||
| Aguda postestreptocócica | mb | n | – | + | n | – | n | n | – | – | + |
| Mesangial IgA | n | n | – | – | n | –/+ | n/a | – | – | – | – |
| Membranoproliferativa | |||||||||||
| Tipo I | b/mb | b | – | – | – | – | n | – | –/+ | –/+ | – |
| Tipo II | m | n/b | – | –/+ | – | n | –/+ | – | – | – | |
| Tipo III | b | n/b | – | – | – | n | – | –/+ | –/+ | – | |
| LES | b | b | + | – | – | – | n | n | – | – | – |
| Rápidamente progresiva | n | n | –/+ | + | + | n | + | – | – | – | |
| Wegener | n | n | – | – | – | –/+ | n | + | – | – | – |
a: alto; AMBG: anticuerpo antimembrana basal glomerular; ANA: anticuerpos antineutrófilos; ANCA: anticuerpos citoplasmáticos antineutrófilos; ASLO: antiestreptolisina O; b: bajo; EGA: estreptococo betahemolítico del grupo A; n: normal; mb: muy bajo; LES: lupus eritematoso sistémico; –: negativo; +: positivo.
Utilizaremos la ecografía para determinar el tamaño renal y las posibles complicaciones. Aunque un tamaño renal normal no excluye insuficiencia renal crónica, ya que pueden estar aumentados sobre su situación basal debido al SN, los riñones pequeños indican fibrosis irreversible, probablemente atrofia renal.
Biopsia renalLos pacientes con hematuria de características glomerulares con presión arterial, función renal normal y proteinuria baja no requieren biopsia renal, a menos que sospechemos una enfermedad sistémica con glomerulonefritis. Las indicaciones absolutas de biopsia renal incluyen un deterioro rápido de la función renal por la sospecha de una glomerulonefritis rápidamente progresiva, insuficiencia renal establecida en la evolución, presencia de proteinuria superior a 1g/1,73m2/día, persistencia de proteinuria y alteraciones inmunológicas no compatibles2,7.
Principales causas etiológicasGlomerulonefritis aguda postestreptocócicaLa glomerulonefritis aguda postestreptocócica (GNAPE) está producida por una infección previa por cepas nefritogénicas de estreptococo betahemolítico del grupo A (EGA)1,2.
EpidemiologíaAunque la GNAPE continúa siendo la causa más común de SN en niños (80%), la mayoría de los casos se registran en los países en vías de desarrollo. Se estima que de los 470.000 nuevos casos anuales, el 97% se registra en estos países. La incidencia anual oscila entre 9,5 y 28,5 por 100.000 individuos10. En los países industrializados, la incidencia ha disminuido en las últimas décadas a 2-4 por 100.000. El riesgo de GNAPE se ha incrementado en pacientes mayores (más de 60 años) y en los niños entre 5 y 12 años. Es muy infrecuente en los niños menores de 3 años. Puede aparecer de forma epidémica o esporádica. La incidencia de GNAPE después de una epidemia de infecciones por EGA es del 5 al 10% de los pacientes con faringitis y del 25% de los pacientes con infecciones cutáneas11,12.
PatogénesisEl mecanismo patogénico más probable de la GNAPE es la formación de inmunocomplejos debido al depósito de antígenos del estreptococo nefritogénico, entre las que se encuentra el serotipo 12, en el glomérulo. Se pensó que esa característica la confería la proteína M, pero se cree que hay 2 posibles antígenos estreptocócicos responsables basados en estudios realizados sobre biopsias10,11. Estos antígenos son: el receptor de la plasmina (NAPlr), una enzima glucolítica que tiene actividad gliceraldehído 3 fosfato dehidrogenasa, y la exotoxina B pirogénica estreptocócica una proteinasa de cisteína catiónica. Ambas proteínas pueden activar la vía alterna del complemento (hallazgocaracterístico de la glomerulonefritis aguda postestreptocócica) y aumentan la expresión de adhesión de las moléculas.
ClínicaLa clínica varía desde pacientes que están asintomáticos, con hematuria microscópica, hasta un SN completo y severo con orinas marrones, proteinuria, hipertensión e insuficiencia renal. Hay un antecedente de infección por EGA en la piel o en la faringe. El periodo de latencia oscila entre una y 3 semanas después de la faringitis, y entre 3 y 6 semanas después de la infección de piel. Algunos pacientes presentan la clínica típica pero no es tan evidente el antecedente de infección por EGA. El edema generalizado aparece en 2 tercios de los pacientes debido a la retención de agua y sodio. En los casos severos, la sobrecarga de líquidos puede producir insuficiencia cardiaca con distrés respiratorio y edema agudo de pulmón. La hematuria macroscópica está presente en un 30-50% de los casos, es de color té o coca-cola y tiene aspecto espumoso. La HTA está presente en un 50-90% de los pacientes y varía desde formas moderadas hasta formas severas. Se debe a la retención de líquidos. La encefalopatía hipertensiva es una complicación infrecuente pero grave. Existen tipos de GNAPE subclínicos, que se caracterizan por hematuria microscópica. Estos pacientes a menudo se detectan durante las epidemias. La GNAPE se asocia a un deterioro variable en la tasa de filtrado glomerular, que se detecta por la elevación de la creatinina. El fallo renal agudo rara vez requiere diálisis12.
Pruebas complementarias específicasAnálisis de orinaHematuria con hematíes dismórficos, con o sin cilindros hemáticos, con grado variable de proteinuria y a menudo piuria. La proteinuria en rango nefrótico es infrecuente, supone un 5% de las presentaciones. Los índices bioquímicos de fracaso renal son de fallo prerrenal porque el sistema renina-angiotensina está intacto; de esta forma, nos encontraremos una fracción de excreción de sodio inferior al 1%.
ComplementoEl 90% de los pacientes tienen el C3 y el CH50 significativamente disminuidos en las 2 primeras semanas del curso de la enfermedad. Los niveles de C2 y C4 están normales o discretamente disminuidos. El C3 y CH50 vuelven a la normalidad entre 4 y 8 semanas después de la presentación. La combinación de un nivel bajo de C3 y C4 normal o discretamente disminuido indican activación de la vía alternativa del complemento.
CultivosSolo el 25% de los pacientes tendrán presente el EGA en la faringe o en la piel, ya que la infección sucede unas semanas antes. En los pacientes con impétigo, hay mayor posibilidad de tener cultivo positivo.
SerologíaLos títulos elevados de anticuerpos contra los productos extracelulares del estreptococo evidencian una infección reciente por EGA. El test de la estreptozima, que mide 5 tipos diferentes de EGA, es positivo en más del 95% de los pacientes con faringitis y del 80% de las infecciones cutáneas incluye los siguientes anticuerpos13: antiestreptolisina O (ASLO); antihialuronidasa; antiestreptoquinasa; antinicotinamida adenina dinucleotidasa y anticuerpos anti-ADNasa B. Estos anticuerpos se pueden medir aisladamente. Después de una infección faríngea el ASLO, anticuerpos anti-ADNasa B, antinicotinamida adenina dinucleotidasa y antihialuronidasa están elevados. En comparación, solo los anticuerpos anti-ADNasa B y antihialirudinasa están elevados después de una infección cutánea. Si solo se usan los títulos de ASLO para hacer el cribado de las infecciones por EGA, puede dar falso negativos en pacientes con infección cutánea. Los títulos de ASLO pueden mitigarse en pacientes con faringitis que han recibido terapia antimicrobiana.
Biopsia renalLa indicación en la fase aguda ya se ha comentado. En el seguimiento, las indicaciones de biopsia renal son: C3 bajo pasadas 8 semanas; C4 descendido de forma mantenida; proteinuria una vez recuperado el SN y hematuria macroscópica persistente. Los hallazgos anatomopatológicos son los siguientes:
- –
Microscopia óptica: glomerulonefritis proliferativa difusa con proliferación endocapilar con infiltrados de neutrófilos. La formación de semilunas es infrecuente y confiere peor pronóstico.
- –
Inmunofluorescencia: depósitos de inmunoglobulina IgG y C3 distribuida en un patrón granular en el mesangio y paredes capilares glomerulares. También puede haber depósitos de IgM, IgA, fibrina y complemento. En los raros casos en los que solo haya depósitos de C3, hay que tener presenta que la evolución puede se peor14.
- –
Microscopia electrónica: depósitos subepiteliales electrón-denso en forma de cúpula (jorobas o humps). Estos depósitos subendoteliales son complejos inmunes que corresponden a IgG y C3 (fig. 4).

El diagnóstico se basa en el antecedente de infección por EGA en la faringe 1-3 semanas o en la piel 3-6 semanas, con hallazgos compatibles con nefritis y disminución de C3 y CH50. El retraso en el diagnóstico es habitual en los casos en que la hematuria es microscópica y en los casos en los que no se tiene claro el antecedente de infección por EGA15. Son excepcionales los casos en los que se diagnostican por la biopsia renal.
Diagnóstico diferencialSe realiza con todas las posibles causas de un SN que expondremos a continuación: GNMP, nefropatía IgA, glomerulonefritis postinfecciosa secundaria a otros agentes y enfermedades sistémicas.
ManejoNo hay un tratamiento específico para la GNAPE. Si se detecta el EGA, se debe tratar para erradicarlo de la faringe. Algunos autores recomiendan el tratamiento sistemático del EGA, independientemente de que se haya aislado en faringe e indican hacer un cribado a los familiares con objeto de erradicarlo. El tratamiento consiste en la restricción de líquidos y sodio. Ya que la mayoría de los pacientes tienen que ingresar, una buena táctica, ya que no sabemos la diuresis previa, consiste en dejar a paciente en dieta absoluta en las 12-24 primeras horas del ingreso; de esa forma, se inicia el balance negativo, continuando posteriormente con ingesta igual a diuresis. Si existe oligoanuria, están indicados los diuréticos de asa, que además reducirán la presión arterial. Se inicia con una dosis de furosemida de 1mg/kg, con un máximo de 40mg16. Si hay hipercaliemia, se utilizan la restricción de potasio de la dieta y las resinas de intercambio iónico. Aunque es infrecuente, los pacientes pueden presentar encefalopatía hipertensiva, en esos casos se debe proceder a reducir la presión arterial, como en las urgencias hipertensivas, de forma lenta. Los pacientes con GNAPE tienen un deterioro variable de la función renal y las técnicas de depuración renal estarán indicadas en los pacientes con fracaso renal agudo severo, sobrecarga de líquidos que no responde a diuréticos y alteraciones hidroelectrolíticas que no responden a medidas habituales.
EvoluciónLa resolución del GNAPE es generalmente bastante rápida. La diuresis típicamente empieza a mejorar en una semana y la creatinina vuelve a valores normales en 2-4 semanas13. Las anomalías urinarias desaparecen progresivamente. La hematuria suele resolverse en 3-6 meses. La proteinuria desaparece durante la recuperación en los casos en los que es más lenta hay que plantearse otra etiología17. La histología mejora a la par que las manifestaciones clínicas, aunque no se suele comprobar porque no se suelen repetir las biopsias. La normalización de la función renal y la resolución de la hematuria antes de que desaparezca la proteinuria reflejan una resolución más lenta de los depósitos subepiteliales comparados con los subendoteliales. Los inmunocomplejos subendoteliales se aclaran rápidamente por las células inflamatorias de la circulación sistémica. No se ve en las biopsias, a menos que sean muy precoces. Los depósitos subepiteliales se separan de las células inflamatorias circulantes por la membrana basal glomerular, por eso está limitado su aclaramiento. En general, el grado de proteinuria se correlaciona con el número de depósitos subepiteliales. La recurrencia de los episodios de GNAPE es rara17,18. Esto se puede deber a la persistencia de anticuerpos frente a los antígenos de estreptococo asociado a nefritis.
PronósticoLa mayoría de los pacientes tienen un excelente pronóstico19. En una serie amplia de 229 niños, un 20% tenía alteración en el análisis de orina (proteinuria y/o hematuria) pero casi todos (92-99%) tenían una función renal normal o mínimamente alterada 5-18 años después de la presentación17. Sin embargo, en algunos casos, como en los adultos, no es tan benigno; pueden desarrollar HTA, proteinuria recurrente e insuficiencia renal de 10 a 40 años después de la enfermedad inicial. Estas manifestaciones se suelen correlacionar con glomeruloesclerosis en la biopsia renal. El mecanismo por el que se produce es hemodinámico; durante el episodio agudo algunos glomérulos se lesionan de forma irreversible y eso provoca hiperfiltración de los restantes. Esta evolución puede mejorar reduciendo la presión arterial, preferiblemente con inhibidores de la enzima convertasa20,21.
Perspectivas futurasLa disponibilidad de una vacuna para los EGA sería deseable para prevenir la enfermedad invasiva y las complicaciones no supurativas. Presumiblemente, una vacuna que erradicara todos los estreptococos del grupo A eliminaría la GNAPE. Actualmente, se dispone de vacuna 26-valente para la mayoría de las cepas reumatogénica pero no incluye las nefritogénicas. La prevención en los países en vías de desarrollo se sigue basando en medidas de salud pública22,23.
Glomerulonefritis membranoproliferativaLa glomerulonefritis membranoproliferativa (GNMP) es una enfermedad glomerular crónica. Es poco frecuente en la infancia pero su importancia radica en que el comienzo puede ser indistinguible de una GNAPE con un SN y, en ocasiones, tiene un curso rápidamente progresivo que termina en insuficiencia renal terminal. El curso clínico prolongado del SN, los niveles de C4 descendidos de forma mantenida más allá de 6 semanas y un deterioro rápido de la función renal nos hará realizar una biopsia renal. El estudio histológico muestra un patrón general de daño glomerular caracterizado en la microscopia óptica por un engrosamiento difuso de la membrana basal glomerular con celularidad aumentada. Tradicionalmente, se han clasificado en 3 tipos según la localización de los depósitos: GNMP tipo I, los depósitos de complemento están en el espacio subendotelial; GNMP tipo II, dentro de la lámina densa de la membrana basal glomerular, con una trasformación electrón densa, y GNMP tipo III, subendoteliales y subepiteliales, con laminación y rotura de la membrana basal. Los depósitos en la tipo I y la tipo III contienen inmunoglobulinas (IgM, IgG) y complemento (C3, C4), mientras que la tipo II o enfermedad de depósitos densos solo contienen C3. Hoy en día, basados en el conocimiento del papel que desempeña el sistema del complemento, nos vemos obligados a cambiar la clasificación y definir, cuando solo encontramos depósitos de C3, la glomerulopatía C3, que tiene un pronóstico malo con evolución a insuficiencia renal terminal pero que, por otro lado, tiene una prometedora opción terapéutica con los anticuerpos monoclonales, que inhiben selectivamente el complejo C5b-9, a pesar de que los ensayos clínicos con eculizumab para esta enfermedad no se hayan realizado todavía9.
Glomerulonefritis mesangial IgALa nefropatía IgA aunque puede debutar igual que la GNAPE como SN pero podemos sospecharla porque si se antecede de proceso respiratorio, el intervalo es más corto, unos 5 días, la duración es menor, el complemento es normal y cursa en brotes, el paciente puede haber tenido un episodio previo o tenerlo posteriormente, a diferencia de la GNPE, que no suele recidivar. Algunos autores refieren aumento de la IgA sérica pero en la práctica clínica en los niños es excepcional, menos de un 5%. La nefropatía IgA cuando comienza como SN agudo y evolución rápidamente progresiva, se ha intentado tratar con pautas inmunosupresoras, aunque el nivel de evidencia es bajo24.
Glomerulonefritis en enfermedades sistémicasLas causas secundarias de glomerulonefritis que se incluyen en el diagnóstico diferencial del SN son: el lupus eritematoso sistémico, la púrpura de Schönlein-Henoch, vasculitis de pequeños vasos (granulomatosis de Wegener y poliangeítis microscópica). La diferenciación radica en las manifestaciones extrarrenales de la enfermedad sistémica y los test de laboratorio. El dato diferencial más marcado será el complemento normal en la púrpura y muy disminuido C3 y C4 en el lupus (figs. 1 y 3). Otras causas posibles son las glomerulonefritis postinfecciosas de otra etiología, las glomerulonefritis asociadas a las endocarditis y a los shunts, y la asociada a la hepatitis B.
La autora declara no tener ningún conflicto de intereses.