





Puntos clave
- •
Muchas inmunodeficiencias primarias (IP) tienen manifestaciones en sangre periférica, fundamentalmente citopenias.
- •
Es necesario prestar atención a la cifra de linfocitos en sangre, sobre todo en los lactantes.
- •
Una cifra de linfocitos < 2.500–3.000/ml en lactantes menores de un año puede ser un signo de inmunodeficiencia combinada grave, incluso en ausencia de otras manifestaciones clínicas.
- •
La citometría de flujo es el método más útil en el diagnóstico de las IP, por su facilidad de realización y rapidez.
- •
El diagnóstico de IP debe estar centrado en el paciente, solicitando las pruebas de laboratorio, paso a paso, de acuerdo con los hallazgos de la historia personal, familiar y de la exploración física.
- •
El escrutinio inicial de IP incluye, casi siempre, hemograma, bioquímica general, cuantificación de inmunoglobulinas y poblaciones de linfocitos T, B y NK.
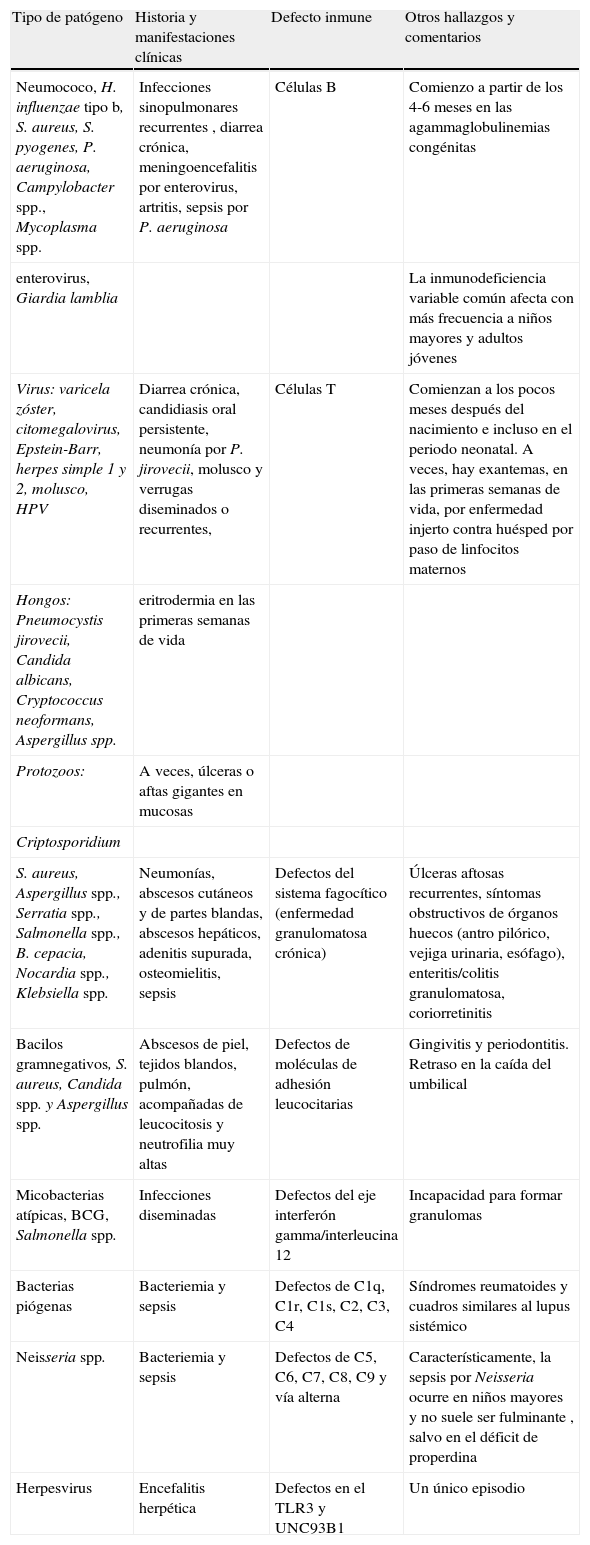
La sospecha de inmunodeficiencia primaria (IP) parte, casi siempre, de un niño o un adulto que presenta infecciones recurrentes, difíciles de tratar, o causadas por microorganismos oportunistas1–5 (tabla 1). Sin embargo, en algunas IP puede haber una infección única en toda la vida del sujeto6,7, manifestaciones autoinmunes8–13 u otros signos o síntomas1,5 más difíciles de relacionar con IP como: úlceras o aftas gigantes de mucosas; rasgos dismórficos faciales; alteraciones cutáneas o del pelo (albinismo, vitíligo, manchas hipercrómicas, petequias, telangiectasias, alopecia); eritrodermias y dermatitis14–20.
Fenotipo clínico de las inmunodeficiencias primarias.
| Tipo de patógeno | Historia y manifestaciones clínicas | Defecto inmune | Otros hallazgos y comentarios |
| Neumococo, H. influenzae tipo b, S. aureus, S. pyogenes, P. aeruginosa, Campylobacter spp., Mycoplasma spp. | Infecciones sinopulmonares recurrentes , diarrea crónica, meningoencefalitis por enterovirus, artritis, sepsis por P. aeruginosa | Células B | Comienzo a partir de los 4-6 meses en las agammaglobulinemias congénitas |
| enterovirus, Giardia lamblia | La inmunodeficiencia variable común afecta con más frecuencia a niños mayores y adultos jóvenes | ||
| Virus: varicela zóster, citomegalovirus, Epstein-Barr, herpes simple 1 y 2, molusco, HPV | Diarrea crónica, candidiasis oral persistente, neumonía por P. jirovecii, molusco y verrugas diseminados o recurrentes, | Células T | Comienzan a los pocos meses después del nacimiento e incluso en el periodo neonatal. A veces, hay exantemas, en las primeras semanas de vida, por enfermedad injerto contra huésped por paso de linfocitos maternos |
| Hongos: Pneumocystis jirovecii, Candida albicans, Cryptococcus neoformans, Aspergillus spp. | eritrodermia en las primeras semanas de vida | ||
| Protozoos: | A veces, úlceras o aftas gigantes en mucosas | ||
| Criptosporidium | |||
| S. aureus, Aspergillus spp., Serratia spp., Salmonella spp., B. cepacia, Nocardia spp., Klebsiella spp. | Neumonías, abscesos cutáneos y de partes blandas, abscesos hepáticos, adenitis supurada, osteomielitis, sepsis | Defectos del sistema fagocítico (enfermedad granulomatosa crónica) | Úlceras aftosas recurrentes, síntomas obstructivos de órganos huecos (antro pilórico, vejiga urinaria, esófago), enteritis/colitis granulomatosa, coriorretinitis |
| Bacilos gramnegativos, S. aureus, Candida spp. y Aspergillus spp. | Abscesos de piel, tejidos blandos, pulmón, acompañadas de leucocitosis y neutrofilia muy altas | Defectos de moléculas de adhesión leucocitarias | Gingivitis y periodontitis. Retraso en la caída del umbilical |
| Micobacterias atípicas, BCG, Salmonella spp. | Infecciones diseminadas | Defectos del eje interferón gamma/interleucina 12 | Incapacidad para formar granulomas |
| Bacterias piógenas | Bacteriemia y sepsis | Defectos de C1q, C1r, C1s, C2, C3, C4 | Síndromes reumatoides y cuadros similares al lupus sistémico |
| Neisseria spp. | Bacteriemia y sepsis | Defectos de C5, C6, C7, C8, C9 y vía alterna | Característicamente, la sepsis por Neisseria ocurre en niños mayores y no suele ser fulminante , salvo en el déficit de properdina |
| Herpesvirus | Encefalitis herpética | Defectos en el TLR3 y UNC93B1 | Un único episodio |
En general, en cualquier infección que curse de manera inexplicable, con manifestaciones clínicas «raras» o acompañada de algunos de los signos anteriores, se debería descartar una IP.
Pruebas de laboratorio en el diagnóstico y la evolución de las inmunodeficiencias primariasLas pruebas de laboratorio para el diagnóstico de las IP pueden dividirse en 4 grandes bloques o niveles: a) pruebas comunes de laboratorio; b) estudios de citometría de flujo (CF); c) pruebas funcionales de los linfocitos, y d) estudios genéticos y moleculares.
Pruebas comunes de laboratorioAunque el diagnóstico final depende muchas veces de estudios inmunológicos, genéticos y moleculares, lo cierto es que en un gran porcentaje de casos se puede hacer una aproximación diagnóstica inicial (la más importante para el pronóstico del niño que padece una IP) con pruebas de laboratorio consideradas «corrientes o habituales».
La sangre constituye la mejor ventana al sistema inmunitario y muchas IP tienen expresión en sangre periférica.
La linfopenia menor de 2.500-3.000 linfocitos/μl en un lactante menor de un año puede ser la primera alerta de una inmunodeficiencia combinada grave (ICG), incluso en ausencia de otras manifestaciones clínicas. Una cifra en este rango obliga a repetir el hemograma para descartar esta grave enfermedad21.
Las citopenias inmunes, afectando a una o más de las 3 series, son frecuentes en varias otras IP22–26. En la inmunodeficiencia variable común (IVC), las citopenias pueden presentarse varios años antes que otras manifestaciones clínicas de la enfermedad. También algunas IP celulares, como el síndrome de DiGeorge, y las deficiencias de adenosín deaminasa (ADA) o nucleótido fosforilasa pueden cursar con citopenias autoinmunes.
En el síndrome de Wiskott-Aldrich (WAS), hay trombocitopenia con plaquetas que tienen un volumen plaquetario disminuido14,15, lo que es diagnóstico de la enfermedad en un varón. De hecho, la trombocitopenia ligada al X puede ser la única manifestación de algunas mutaciones del gen (WAS)14–16. Más raramente, otras mutaciones asociadas a una ganancia de proteína dan lugar a la neutropenia crónica ligada al X14–16,27.
Por último, la cuantificación de inmunoglobulinas es una prueba sencilla y esencial para el diagnóstico de las inmunodeficiencias humorales.
Caracterización de las inmunodeficiencias primarias por citometría de flujo28–30La citometría de flujo es la técnica más útil en el estudio de la mayoría de las IP, ya que permite evaluar las poblaciones y subpoblaciones celulares, así como identificar moléculas de la membrana y del citosol, e incluso valorar la función anormal de una proteína.
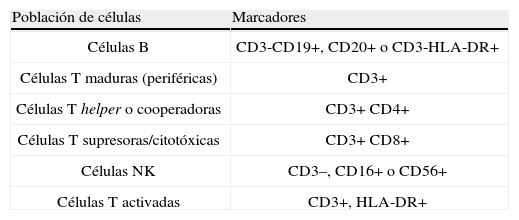
Como escrutinio de las IP, es útil realizar un inmunofenotipo de las poblaciones celulares de la sangre mediante la identificación de marcadores celulares de superficie mediante CF (tabla 2). Con ello se logra la presunción diagnóstica de la mayoría de IP de células T y células B, que posteriormente se confirma, mediante otros estudios (tabla 3).
Poblaciones y subpoblaciones linfocitarias por citometría de fujo en el escrutinio de inmunodeficiencias primarias.
| Población de células | Marcadores |
| Células B | CD3-CD19+, CD20+ o CD3-HLA-DR+ |
| Células T maduras (periféricas) | CD3+ |
| Células T helper o cooperadoras | CD3+ CD4+ |
| Células T supresoras/citotóxicas | CD3+ CD8+ |
| Células NK | CD3–, CD16+ o CD56+ |
| Células T activadas | CD3+, HLA-DR+ |
NK: natural killer.
Diagnóstico de las inmunodeficiencias primarias por citometría de flujo.
| Enfermedad | Diagnóstico por citometría de flujo |
| Agammaglobulinemia congénita ligada al X | Tirosina cinasa de Bruton en monocitos y plaquetas |
| Síndrome de hiper-IgM ligado al X | CD40L (CD154) en las células T activadas |
| Síndrome de hiper-IgM tipo 3 | CD40 en las células B o en los monocitos |
| Alteraciones en la inmunodeficiencia variable común | ICOS (células T activadas), BAFF, TACI y CD19 (en linfocitos B)Células switch memory (CD27 + IgD–IgM–) y linfocitos B CD21low |
| Síndrome WHIM (warts o verrugas, hipogammaglobulinemia, infecciones y mielocatexis) | CXCR4 (linfocitos T) |
| Inmunodeficiencia combinada grave por déficit de cadena gamma común | CD132 en células T activadas |
| Síndrome del linfocito desnudo | |
| Tipo I | Expresión de MCH-I en monocitos |
| Tipo II | Expresión de MCH-II en células B y células T activadas |
| Deficiencia de CD25 | CD25 (IL2Rα) |
| Inmunodeficiencia con enteropatía ligada al X | FOXP3 en células T reguladoras (CD4+, CD25+, FOXP3+) |
| Síndrome de Wiskott Aldrich, trombocitopenia crónica ligada al X, neutropenia crónica ligada al X | Proteína WAS |
| Enfermedad granulomatosa crónica | Producción de radicales libres del oxígeno (explosión o burst de oxígeno) |
| Deficiencia de moléculas de adhesión leucocitarias tipo I | Expresión de CD18, CD11a y CD11b en los leucocitos |
| Deficiencia de moléculas de adhesión leucocitarias tipo II | Expresión de CD15 en neutrófilos y linfocitos |
| Deficiencia del receptor 1 del IFN-γ | IFN-γR1 |
| Deficiencia del receptor 2 del IFN-γ | IFN-γR2 |
| Deficiencia del receptor β1 de la IL-12 y de la IL-23 | IL-12Rβ1 |
| Linfohistiocitosis hemofagocítica familiar | Expresión de perforina en los linfocitos CD8+ y ensayo de degranulación del CD107a |
| Síndrome linfoproliferativo ligado al X | SAP (SH2D1A) |
| Síndrome linfoproliferativo ligado al X por deficiencia del inhibidor de la apoptosis | XIAP |
IL: interleucina; IFN-γ: interferón gamma. SAP: proteína asociada a SLAM; XIAP: deficiencia proteina que inhibe la apoptosis ligada por X).
Esta IP, la más frecuente de las formas de las ICG (50%), se produce por mutaciones en el gen que codifica la cadena común (gC o CD132), que forma parte de los receptores de las interleucinas (IL)-2, IL-4, IL-7, IL-9, IL-15 e IL-21. La IL- 7 es esencial para el desarrollo de los linfocitos T, mientras que la IL-15 lo es para el desarrollo de las células NK. Por tanto, los individuos con esta mutación carecen de ambos tipos celulares, pero sí tienen células B (fenotipo T–, B+, NK–). El diagnóstico definitivo se hace demostrando, por CF, la expresión anormal o ausente de la cadena gC (CD132). El estudio genético permite identificar la mutación causante de la enfermedad.
Inmunodeficiencia combinada grave autosómica por mutación de Jak-3La enzima Janus cinasa 3 (Jak-3) es una tirosincinasa que se asocia a la porción citoplasmática de los receptores de varias IL (entre otras, IL-2, IL-4, IL-7, IL-9, IL-15 e IL-21) formando, por tanto, parte de la vía de activación celular. La mutación de Jak-3 da lugar a una ICG con idéntico fenotipo al de la ICG ligada al X (T–, B+, NK–), pero menos frecuente y con herencia autosómica recesiva.
Inmunodeficiencia combinada grave por mutaciones en el receptor de la interleucina 7 (IL-7Ra)La unión de la IL-7 con su receptor es esencial para el desarrollo de los linfocitos T. La ausencia de este receptor da lugar a una inmunodeficiencia con fenotipo T–, B+, NK+.
Inmunodeficiencia T–, B-NK+ por mutaciones en los genes RAG1, RAG2, artemis y ligasa IVLos productos de los genes anteriores intervienen en el mecanismo de recombinación (rearrangement) del ADN de la célula B y T, que genera la extraordinaria diversidad de inmunoglobulinas y receptores de la célula T (TCR), necesaria para enfrentarse a cualquier antígeno potencial.
La mutación en cualquiera de estos genes afecta a tanto al desarrollo de los linfocitos T como de los B, y da lugar a una ICG grave con fenotipo T–, B– y NK+. Las mutaciones en el complejo RAG1/RAG2 causan el 20% de ICG en Europa.
Inmunodeficiencia combinada grave por déficit de adenosín deaminasaLa deficiencia de ADA hace que se acumule un metabolito tóxico para los linfocitos, dando lugar a una profunda linfopenia (habitualmente menos de 500/μl). El fenotipo linfocitario de esta enfermedad es T–, B–, NK–.
Síndrome de OmennEn algunos pacientes con mutaciones hipomórficas (que no causan la pérdida de función total de la proteína codificada) de Artemis, RAG1/RAG2 e IL-7R, IL-2R, ADA, ADN-ligasa 4 y RN-asa mitocondrial se produce una inmunodeficiencia con aumento de secreción de citocinas, que causan fenómenos autoinmunes e inflamación alérgica. El aumento de citocinas se debe a una expansión clonal de una población de células T, que están anormalmente activadas (como lo demuestra el aumento de su expresión de HLA DR+). Además tienen un repertorio contraído del TCR, que se puede demostrar por CF. Clínicamente, los pacientes presentan eritrodermia, hepatoesplenomegalia, diarrea e infecciones recurrentes. Hay eosinofilia en sangre periférica y aumento de IgE.
Síndrome de DiGeorgeEl diagnóstico de las diferentes formas de síndrome de DiGeorge exige una valoración cuidadosa del fenotipo físico y la demostración de la deleción del 22q11.2. La CF ayuda a establecer la gravedad y el grado de inmunodeficiencia, determinando las subpoblaciones de los linfocitos T, así como función de los mismos mediante la respuesta proliferativas a mitógenos.
Otras inmunodeficiencias celularesEn la tabla 3, se exponen otras IP celulares que pueden diagnosticarse por CF.
Inmunodeficiencias humorales1–4,31,32La CF es también primordial en el estudio de las IP humorales, ya que permite, de forma muy sencilla, hacer una primera aproximación diagnóstica al saber si el paciente tiene o no células B. Los marcadores que identifican las células B por CF son: a) el CD19 que es el marcador que identifica a todas las células B, en cualquiera de sus estadios evolutivos, excepto en el de célula plasmática; b) el CD20 que aparece después del anterior en la diferenciación del linfocito B; c) el CD21 que es un marcador de activación de los linfocitos B maduros, y d) los HLA-DR, expresados de forma constitutiva por los linfocitos B (los linfocitos T solo lo hacen cuando están activados).
Agammaglobulinemias congénitasAbarcan tanto la forma ligada al X por déficit de la tirosina cinasa de Bruton (Btk), como las formas autosómicas. Todas ellas se caracterizan, además de por agammaglobulinemia (cifras de IgG, IgA e IgM por debajo de 2 desviaciones estándar [DE] de los valores normales) por poseer un fenotipo CD3+, CD19–, CD20-CD21– y HLA-DR–, es decir, una ausencia de células B (menos del 2% de los linfocitos).
La mayoría de las agammaglobulinemias congénitas responde a la deficiencia de Btk, cuya ausencia de expresión puede demostrarse en plaquetas y monocitos por CF. La ausencia de esta proteína es sinónima de la enfermedad, pero su presencia no descarta el diagnóstico, ya que la proteína expresada puede ser funcionalmente anormal.
Síndromes de hiper-IgM o deficiencias en la recombinación para el cambio de isotipoTras el estímulo antigénico, la célula B en reposo, que expresa IgM e IgD en su superficie, sufre una expansión clonal con secreción de IgM. Posteriormente, se produce el cambio de isotipo o switching (se forman células plasmáticas que secretan IgG o IgA en lugar de IgM), maduración de la afinidad de los anticuerpos (por hipermutación somática) y producción de células de la memoria. Para que se produzcan estos eventos, es necesaria la unión del CD40L del linfocito T helper (TH) activado y el CD40 del linfocito B. Los pacientes en los que esta conexión fracasa, por mutaciones en el CD40 o en el CD40L, tienen una IgG e IgA bajas con IgM alta o normal, al no producirse el cambio de isotipo o switching. Otros defectos genéticos de la vía de activación del CD40-CD40L son el modulador esencial NF-kB, la uracil-ADN-glucosidasa, la activación inducida de la citidin deaminasa, IKBA y PMS2.
Además de las alteraciones de la inmunidad humoral, los defectos en el CD40L tienen también profundas alteraciones de la célula T e infecciones oportunistas.
El síndrome de Hiper-IgM más frecuente (aproximadamente el 75% de los casos) es la forma ligada al X por deficiencia del CD40L (síndrome HIGEX). En esta forma falta la expresión del CD40L (CD154) en los TH previamente activadas (no se expresa en las células en reposo) con ionóforos del calcio más forbol-miristol acetato (PMA).
También por CF se determina el receptor CD40 en las células B (CD19 o CD 20).
Las células B de memoria vienen identificadas por la expresión de la molécula CD27 en su superficie. Por tanto, su fenotipo es CD19+, CD27+. Las células de memoria sin cambio de isotipo (non-switching) son CD19+, CD27+, IgM+ y IgD+ (expresan en su superficie las inmunoglobulinas IgM e IgD), mientras que la célula de memoria con cambio de isotipo (célula switch memory) tiene el fenotipo CD19+, CD27+, IgM– e IgD–. Estas células están disminuidas en el síndrome HIGEX (pero no en las formas autosómicas del síndrome), en algunos subgrupos de inmunodeficiencia variable común y de síndrome linfoproliferativo autoinmmune28.
Inmunodeficiencia variable común9,13,26,33,34Esta IP se caracteriza por disminución significativa de los niveles de IgG (caída por debajo de 2DS) e IgA y/o IgM, con producción disminuida o ausente de anticuerpos, un número de células B circulantes (> 2%) y exclusión de otras causas de hipogammaglobulinemia.
Como en todas las hipogammaglobulinemias, el primer paso es determinar, por CF, las subpoblaciones linfocitarias T y B (tabla 2) para demostrar que no se trata de una agammaglobulinemia congénita.
La producción de anticuerpos específicos se mide determinando las isohemaglutininas u otros anticuerpos frente a antígenos a los que el individuo ha estado expuesto (vacunas tétanos, difteria, hepatitis B, hepatitis A, rubéola, sarampión) o vacunado al sujeto con distintas vacunas y demostrando el incremento de títulos de anticuerpos entre una muestra de suero prevacunal y posvacunal.
La CF permite también definir subgrupos de la IVC9,13,34. Los pacientes que tienen menos del 2% de células B de memoria con cambio de isotipo (CD19+, CD27+, IgM–, IgD –) tienen mayor riesgo de esplenomegalia, granulomatosis y niveles más bajos de inmunoglobulinas30. Otro subgrupo de IVC es el constituido por pacientes con expansión de una población de linfocitos B (caracterizada por una expresión baja de CD21 y de CD38, y un aumento de la expresión de CD19) conocida como CD21low. Un porcentaje de CD21low superior al 10% de los linfocitos B se asocia, más que ningún otro parámetro, a esplenomegalia y citopenias autoinmunes9,30.
También es posible identificar por CF los defectos genéticos de algunas formas de IVC como ICOS, CD19, CD21 y BAFF-R26,33,35.
Inmunodeficiencias primarias que afectan a la inmunidad innataEnfermedad granulomatosa crónica19,20Mediante citometría de flujo puede determinarse la producción de radicales derivados del oxígeno para el diagnóstico de las diferentes formas de enfermedad granulomatosa crónica, en las que existe una incapacidad para generar radicales libre del oxígeno, por defectos en alguno de los componentes del sistema NADPH. El llamado test de la explosión o burst de oxígeno mide la producción de estos radicales mediante citometría de flujo utilizando un colorante intracelular —la dihidrorodamina 123—, que se convierte en un colorante fluorescente al activar los granulocitos con PMA (formol-miristol-acetato). Las células de los sujetos con la enfermedad no presentan fluorescencia o está muy disminuida, a diferencia de los sujetos normales. En las madres portadoras hay 2 grupos de poblaciones celulares.
Defectos del eje interleucina-12/interferón gamma6,7Esta vía implica los receptores del interferón gamma (INF-γ), IFN-gR1 e IFN-gR2, la IL-12 (IL12-p40) y el receptor de la IL-12 (IL-12Rb1). La IL-12, producida por las células dendríticas, activa los linfocitos CD4+ y células NK para que produzcan IFN-g, que, a su vez, activa los macrófagos y estimula a las células TCD4+, dando lugar a una respuesta Th1. Las alteraciones de esta vía dan lugar a infecciones por organismos intracelulares, como infecciones diseminadas por micobacterias atípicas (Mycobacterium avium sobre todo), BCG, y Salmonella spp. La ausencia de las moléculas anteriores se demuestra por CF.
El IFN-g activa a célula CD4+ a través de la fosforilización de un activador de la transcripción llamado STAT1, que facilita la transcripción de varios genes de activación, mientras que la IL-12 activa el linfocito CD4+ por la vía de STAT4. La falta de fosforilización de STAT 1 o STAT4 demostrada por CF sugiere defectos en la vía del IFN-g y la IL-12, respectivamente.
Inmunodeficiencias con disregulación inmuneLinfohistiocitosis eritrofagocítica familiarHay 3 defectos genéticos conocidos que se asocian a esta IP: el déficit de perforina, la mutación en Munc-13 y la mutación de la sintaxina 11.
El déficit de perforina, que corresponde al 20-30% de los casos de esta enfermedad, puede demostrarse, por CF, mediante tinción intracelular de las células NK (CD16+/CD57+) y linfocitos citotóxicos (CD8+).
La expresión de CD107a por las NK siguiendo a su estimulación predice los defectos de Munc-13 y sintaxina 11, y puede ser utilizada como cribado.
Síndrome linfoproliferativo autoinmuneEste síndrome se caracteriza por linfoproliferación (linfadenopatía y esplenomegalia crónicas), hipergammaglobulinemia y fenómenos autoinmunes (los más frecuentes son las citopenias)36. Se debe a defectos en las vías que conducen a la apoptosis celular, encargada de mantener la homeostasis linfocitaria. La forma más frecuente obedece a deficiencia de FAS (CD95/APO1). Otras formas menos habituales son las mutaciones en Fas ligando (FASL) y las caspasas intracelulares 8 y 10, todas ellas implicadas en la vía de la apoptosis mediadas por FAS.
En todas estas formas es muy característico el aumento de las llamadas células T dobles negativas, que no expresan la molécula CD4 ni CD8 (células TCRab+, CD4–, CD8–). Cuando estas células suponen más de un 1,5% de los linfocitos totales o > 2,5% de los linfocitos CD3+, la posibilidad de esta enfermedad es muy alta. También por CF, mediante diversos métodos, puede evaluarse la apoptosis mediada por Fas.
Otras inmunodeficiencias primarias que se puede estudiar mediante CFLos defectos de moléculas de adhesión tipo 1 (LAD-1) se produce por mutaciones en el gen que codifica la cadena b (CD18) de las b-integrinas leucocitarias. La ausencia de esta cadena imposibilita el transporte a la superficie celular de otras cadenas (CD11a, CD11b, CD11c) que forman parte de las b-integrinas. La disminución de expresión de la molécula CD11b en los granulocitos en reposo o estimulados, demostrada mediante CF, sirve para realizar el diagnóstico de la enfermedad, al tiempo que permite establecer grupos pronósticos, según la intensidad de la disminución.
En la tabla 3 se exponen otras IP a cuyo diagnóstico se puede llegar por CF.
Respuestas proliferativas linfocitarias a mitógenos y antígenosSe utilizan para evaluar la función linfocitaria, provocando «in vitro» la activación y la proliferación de los linfocitos mediante diferentes moléculas, que simulan la activación natural del linfocito TH37 (fig. 1). La función y el punto donde actúan los mitógenos, que se usan solos o en combinación con otros, se exponen en la tabla 4.
![Activación de linfocito T helper. La célula presentadora de antígeno (APC) fagocita y elabora, digiriendo el antígeno, un pequeño péptido, que es presentado mediante los HLA-II al TCR (receptor específico del linfocito T) con sus 2 cadenas a y b, pertenecientes a la superfamilia de las inmunoglobulinas, y que está en conexión con el complejo CD3 formado, a su vez, por varias moléculas. La presentación del antígeno activa una serie de enzimas (muchas de ellas cinasas) que fosforilizan diferentes proteínas y finalmente activan a la fosfolipasa C (PLCg). Esta enzima hidroliza los fosfolípidos de la membrana (fosfatidil insositol bifosfato o PIP2 en inositol trifosfato [IP3] y diacilglicerol [DAG]). El IP3 pasa al citosol y libera Ca++ del retículo endoplásmico, aumentando la concentración citosólica del Ca++. El DAG queda en la membrana celular y estimula la fosfocinasa C (PKC) que pasa al citosol. El aumento de Ca++ citosólico y la activación de la PKC activan a su vez diferentes factores de transcripción (NF-κB, NFAT, AP1) que se traslocan al núcleo y activan la transcripción de diferentes genes, fundamentalmente los genes de la IL-2 y su receptor, cuyo resultado final es la expansión clonal específica del linfocito T helper que ha reconocido al antígeno. En la activación del linfocito T intervienen, además, otras moléculas —llamadas moléculas coestimulatorias— que potencian las reacciones anteriores. Dos de las más importantes son la B7, que aparece en la APC, y la CD28, que se expresa en el linfocito T helper y reconoce a la anterior. Estas moléculas potencian la activación del linfocito T.](https://static.elsevier.es/multimedia/16962818/0000001100000005/v1_201310160028/S1696281813701495/v1_201310160028/es/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNeGnnX4Hqh7LROCAxmf2LVNvrMylODO3Qx/VrPDaq3iDKBeGXx2Lg/p+fxMzrZwyf4lS8FM/7MQWVSvN1LU9w6KL+NY5MydTPdc2QpvjI3YzaWiDcdgkQ04SfYAlZBQ6mS0KQpObJiwWW+PZSWj5GKR+6fay8HRgiXbO2vvbYtH81JfTm36O84IbNTAG77ZYCF0QHTw6wVwWOMAqkYxNmn63wc58qEl9Nb6D57EKul5gVQ4RVg0rjf5hO1tSzDU6GI= "Activación de linfocito T helper. La célula presentadora de antígeno (APC) fagocita y elabora, digiriendo el antígeno, un pequeño péptido, que es presentado mediante los HLA-II al TCR (receptor específico del linfocito T) con sus 2 cadenas a y b, pertenecientes a la superfamilia de las inmunoglobulinas, y que está en conexión con el complejo CD3 formado, a su vez, por varias moléculas. La presentación del antígeno activa una serie de enzimas (muchas de ellas cinasas) que fosforilizan diferentes proteínas y finalmente activan a la fosfolipasa C (PLCg). Esta enzima hidroliza los fosfolípidos de la membrana (fosfatidil insositol bifosfato o PIP2 en inositol trifosfato [IP3] y diacilglicerol [DAG]). El IP3 pasa al citosol y libera Ca++ del retículo endoplásmico, aumentando la concentración citosólica del Ca++. El DAG queda en la membrana celular y estimula la fosfocinasa C (PKC) que pasa al citosol. El aumento de Ca++ citosólico y la activación de la PKC activan a su vez diferentes factores de transcripción (NF-κB, NFAT, AP1) que se traslocan al núcleo y activan la transcripción de diferentes genes, fundamentalmente los genes de la IL-2 y su receptor, cuyo resultado final es la expansión clonal específica del linfocito T helper que ha reconocido al antígeno. En la activación del linfocito T intervienen, además, otras moléculas —llamadas moléculas coestimulatorias— que potencian las reacciones anteriores. Dos de las más importantes son la B7, que aparece en la APC, y la CD28, que se expresa en el linfocito T helper y reconoce a la anterior. Estas moléculas potencian la activación del linfocito T.")
Activación de linfocito T helper. La célula presentadora de antígeno (APC) fagocita y elabora, digiriendo el antígeno, un pequeño péptido, que es presentado mediante los HLA-II al TCR (receptor específico del linfocito T) con sus 2 cadenas a y b, pertenecientes a la superfamilia de las inmunoglobulinas, y que está en conexión con el complejo CD3 formado, a su vez, por varias moléculas. La presentación del antígeno activa una serie de enzimas (muchas de ellas cinasas) que fosforilizan diferentes proteínas y finalmente activan a la fosfolipasa C (PLCg). Esta enzima hidroliza los fosfolípidos de la membrana (fosfatidil insositol bifosfato o PIP2 en inositol trifosfato [IP3] y diacilglicerol [DAG]). El IP3 pasa al citosol y libera Ca++ del retículo endoplásmico, aumentando la concentración citosólica del Ca++. El DAG queda en la membrana celular y estimula la fosfocinasa C (PKC) que pasa al citosol.
El aumento de Ca++ citosólico y la activación de la PKC activan a su vez diferentes factores de transcripción (NF-κB, NFAT, AP1) que se traslocan al núcleo y activan la transcripción de diferentes genes, fundamentalmente los genes de la IL-2 y su receptor, cuyo resultado final es la expansión clonal específica del linfocito T helper que ha reconocido al antígeno.
En la activación del linfocito T intervienen, además, otras moléculas —llamadas moléculas coestimulatorias— que potencian las reacciones anteriores. Dos de las más importantes son la B7, que aparece en la APC, y la CD28, que se expresa en el linfocito T helper y reconoce a la anterior. Estas moléculas potencian la activación del linfocito T.
Mitógenos y antígenos utilizados para valorar la función linfocitaria.
| Mitógeno o antígeno | Función y significado |
| Superantígenos (enterotoxinas estafilocócicas A y C) | Se unen, sin procesamiento, al borde lateral del HLA-II de la célula presentadora de antígeno, y del complejo TCR-CD3. Estimula a un número mucho mayor de linfocitos T que los antígenos (hasta un 20%) |
| Anti-CD3 | Estimula el receptor CD3, saltándose el reconocimiento del antígeno, e inicia la cascada que conduce a la activación de la célula T |
| Lectinas: fitohemaglutinina y concanavalina | Aglutinan azúcares de moléculas como TCR-CD3, CD4,CD8, CD2, CD5, CD43 y activan al linfocito T de forma independiente al TCR |
| Ionóforos del Ca++ (ionomicina) | Actúan de manera similar al inositol trifosfato, aumentando el Ca++ intracelular |
| PMA | Tiene el mismo mecanismo de actuación que la PKCEl uso combinado de ionomicina y PMA activa el linfocito T, independientemente de CD3 y CD2 |
| CD28 | Es una molécula coestimulatoria. Estabiliza los transcriptos de ARN de la IL-2, IFN-γ, GM-CSF, TNF-a y potencia a otros mitógenos como CD3, CD2, lectinas y PMA |
| CD26 y CD69 | Moléculas coestimulatorias. Potencia la proliferación de los linfocitos T estimulados por PMA |
| IL-2 | Corrige la proliferación den los defectos de IL-2 con expresión normal de los receptores de la IL-2 |
| Mitógeno Pokeweed | Mitógeno de la célula B, dependiente de la célula T. Explora función de células B y T, y la cooperación entre ellas |
| Anti-CD2 | El CD2 o T11 es el receptor de los hematíes de carnero de los linfocitos T. Es el marcador Pan-T por excelencia. En anti-CD2 hace proliferar al linfocito T por una vía alternativa al TCR-CD3 |
TNF: factor de necrosis tumoral; HLA: antígeno de histocompatibilidad de clase A; IFN-V: interferón gamma; Ig: inmunoglobulina. IL: interleucina; PMA: forbol miristol acetato; TCR: receptores de la célula T; TNF-α: interferón alfa.
Para estudiar la respuesta del linfocito THa los mitógenos y antígenos, se obtienen células mononucleares de sangre periférica en heparina o EDTA, que se colocan en un medio de cultivo con nucleótidos, algunos de ellos con una base marcada, como la timidina tritiada (TT). El grado de activación o proliferación de linfocitos «in vitro» en respuesta a un estímulo (mitógeno o antígeno) se puede evaluar mediante la medida de la incorporación de la TT al ADN que se sintetiza de novo por las células en proliferación. La proliferación de las células del paciente se compara con la del control sano.
Una proliferación normal tras el uso de un mitógeno concreto implica que toda la vía de activación, desde el punto donde actúa el mitógeno hasta la síntesis de IL-2 y su receptor, que son los estadios finales de la activación y proliferación celular final, es normal. Por el contrario, una proliferación disminuida significa algún trastorno de la vía a partir del punto de actuación del mitógeno. Así, utilizando diversos mitógenos o antígenos, se puede llegar a conocer dónde se encuentra el trastorno funcional del linfocito TH.
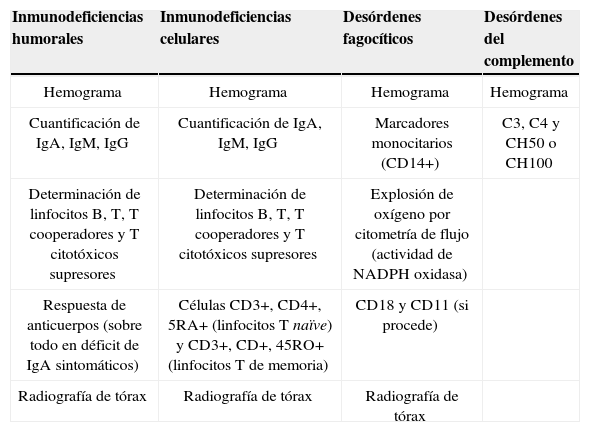
Cribado de inmunodeficiencia primaria5El escrutinio inicial de laboratorio de IP debe estar centrado en el paciente, dependiendo de la historia personal y familiar y de las manifestaciones clínicas (tabla 1). Después debe establecerse un protocolo paso a paso hasta llegar al diagnóstico mediante otras pruebas de CF (tablas 2 y 4), test de proliferación linfocitaria (tabla 5) y estudios de genética y biología molecular.
Pruebas a realizar según el tipo de inmunodeficiencia que se sospecha.
| Inmunodeficiencias humorales | Inmunodeficiencias celulares | Desórdenes fagocíticos | Desórdenes del complemento |
| Hemograma | Hemograma | Hemograma | Hemograma |
| Cuantificación de IgA, IgM, IgG | Cuantificación de IgA, IgM, IgG | Marcadores monocitarios (CD14+) | C3, C4 y CH50 o CH100 |
| Determinación de linfocitos B, T, T cooperadores y T citotóxicos supresores | Determinación de linfocitos B, T, T cooperadores y T citotóxicos supresores | Explosión de oxígeno por citometría de flujo (actividad de NADPH oxidasa) | |
| Respuesta de anticuerpos (sobre todo en déficit de IgA sintomáticos) | Células CD3+, CD4+, 5RA+ (linfocitos T naïve) y CD3+, CD+, 45RO+ (linfocitos T de memoria) | CD18 y CD11 (si procede) | |
| Radiografía de tórax | Radiografía de tórax | Radiografía de tórax |
IL: interleucina; IFN-g: interferón gamma; PKC: fosfocinasa C; PMA: forbol-miristol-acetato; TCR: receptores de la célula T; TNF-α: factor de necrosis tumoral alfa
Los autores declaran no tener ningún conflicto de intereses.