




Puntos clave
- •
La enfermedad de células falciformes (ECF) es multisistémica. El pediatra debe conocerla para prevenir y reconocer las complicaciones agudas que pueden ser mortales.
- •
Los pacientes con ECF tienen una asplenia funcional y un riesgo alto de sepsis por neumococo. La profilaxis con penicilina oral y vacunación, así como el tratamiento empírico frente a este germen en los episodios febriles, reducen la mortalidad infantil por ECF.
- •
El secuestro esplénico puede ocasionar un shock hipovolémico y ser mortal en menos de una hora si no se transfunde urgentemente.
- •
El papel del pediatra es esencial en la prevención de nuevos casos y en el diagnóstico precoz mediante estudio familiar y consejo genético.
- •
Para prevenir crisis vasooclusivas los pacientes deben estar bien hidratados, evitar el frío y el calor, y hacer ejercicio moderado.
- •
Se debe evitar una hemoglobina superior a 10 g/dl postransfusión para evitar un aumento en la viscosidad sanguínea y del riesgo de vasooclusión.
La enfermedad de células falciformes (ECF) o drepanocitosis es una enfermedad multisistémica que cursa con complicaciones agudas y lesiones crónicas en diversos órganos. Es una de las enfermedades monogénicas más frecuentes en el mundo y se debe a una mutación en el gen beta de la hemoglobina. Afecta predominantemente, pero no de forma exclusiva, a individuos de origen africano1. Debido a la inmigración, el número de casos en España ha aumentado mucho en los últimos años2. En la actualidad, es imprescindible que el pediatra conozca la ECF para reducir la morbimortalidad infantil. Será el pediatra quien, con frecuencia, diagnostique a estos niños, dé un consejo genético, inicie la profilaxis infecciosa y derive a un centro hospitalario ante la sospecha de alguna complicación aguda, que puede ser muy grave. Esta revisión trata de la fisiopatología de esta enfermedad, su diagnóstico, sus manifestaciones clínicas y tratamiento, con especial énfasis en el papel del pediatra generalista.
FisiopatologíaLa drepanocitosis es la primera enfermedad en la que se conoció su base molecular3. Se debe a una mutación en el gen de la cadena beta de la hemoglobina que produce la sustitución de un aminoácido (glutamina por valina) en dicha cadena. Este cambio altera las propiedades físico-químicas de esta molécula, denominada hemoglobina S (HbS), de manera que cuando está desoxigenada forma polímeros intracelulares y el hematíe adopta forma alargada o de hoz (sickle en inglés)4. Los hematíes falciformes o drepanocitos son rígidos y pierden la elasticidad necesaria para que la circulación sanguínea sea fluida. Estos hematíes se destruyen con facilidad, por lo que una de las manifestaciones clínicas es la anemia hemolítica. Además, los hematíes falciformes tienden a formar agregados en las vénulas poscapilares que obstruyen la circulación (vasooclusión) e interaccionan con el endotelio, leucocitos y plaquetas, produciendo isquemia y lesiones crónicas en diversos órganos. La hemólisis tiene un papel central en algunas de las complicaciones de esta enfermedad, como son la hipertensión pulmonar, las úlceras maleolares y el priapismo5. La hemólisis intravascular da lugar a la liberación de hemoglobina libre y a arginasa con lo que disminuye la disponibilidad de óxido nítrico (NO). La disminución de NO inhibe la vasodilatación y estimula la activación y proliferación del endotelio.
En algunos pacientes predominan las complicaciones derivadas de la hemólisis; suelen ser aquellos con mayor anemia basal y más datos de hemólisis (reticulocitosis más alta, lactatodeshidrogenasa más elevada). En otros pacientes predominan las complicaciones por vasooclusión, como son las crisis de dolor y el síndrome torácico agudo. Estos últimos suelen tener hematocritos más altos.
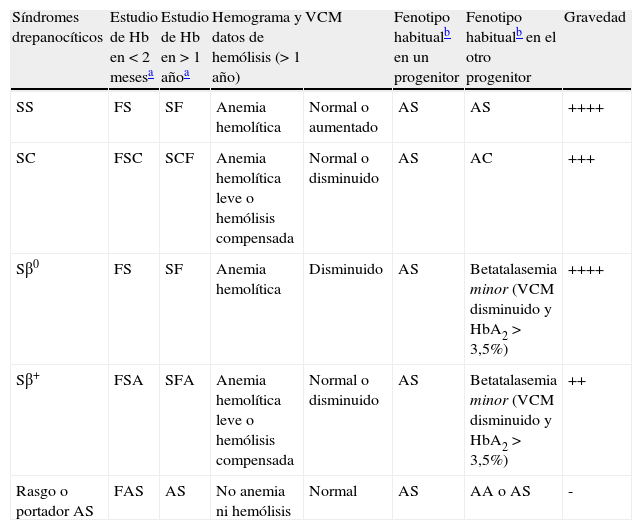
Herencia y variantesLa ECF se transmite de forma autosómica recesiva. Los individuos heterocigotos (AS) son portadores asintomáticos. La enfermedad tiene lugar cuando se hereda de forma homocigota el gen S de ambos progenitores (SS) o bien de forma doble heterocigota, junto con otra hemoglobinopatía como HbC (SC), D (SD) y otras. Si se hereda de un progenitor el gen S y de otro un gen de betatalasemia, el individuo tendrá una drepanocitosis doble heterocigota beta-S (Sβ0 o Sβ+). Las mutaciones del gen beta pueden acarrear la incapacidad absoluta de producir una cadena beta normal (β0) o resultar en una reducción de su producción (β+). Tanto en la forma homocigota como en la doble heterocigota, la consecuencia es un predominio de HbS en lugar de HbA6. En general, las formas SC y la Sβ+ suelen ser más leves que las SS y Sβ0 (véase la tabla 1).
Lectura rápida
La enfermedad de células falciformes (ECF) es una hemoglobinopatía de herencia autosómica recesiva. Los individuos heterocigotos son portadores sanos (AS) y los homocigotos (SS) o doble heterocigotos (SC, S-betatalasemia) presentan la enfermedad. En España, ha aumentado mucho su incidencia debido a la inmigración, pues la ECF afecta principalmente a individuos procedentes de África, Arabia Saudí, Grecia, India y América.
FisiopatologíaSe debe a una mutación en el gen beta de la hemoglobina que altera las propiedades físico-químicas de la molécula, denominada hemoglobina S, de modo que cuando está desoxigenada forma polímeros intracelulares y el hematíe adopta forma alargada o de hoz. Los hematíes falciformes son rígidos y pierden la elasticidad necesaria para que la circulación sanguínea sea fluida. Se destruyen con facilidad y, además, forman agregados en las vénulas poscapilares (vasooclusión) que obstruyen la circulación produciendo isquemia.
El diagnóstico se puede realizar por morfología eritrocitaria y confirmar por estudio de hemoglobinas. El estudio molecular es útil en el diagnóstico prenatal. Se puede realizar un diagnóstico precoz mediante cribado de sangre de talón en el neonato que permite instaurar medidas preventivas de la sepsis neumocócica, educación sanitaria y consejo genético.
El fenotipo de los niños con ECF es muy variable, algunos presentan pocas complicaciones y otros precisan varios ingresos al año. Las principales manifestaciones clínicas se deben a los fenómenos vasooclusivos, a la hemólisis y al hipoesplenismo funcional. Estos pacientes presentan complicaciones agudas y otras crónicas por la vasculopatía progresiva que afecta a distintos órganos.
Hipoesplenismo funcional: se debe a fenómenos vasooclusivos en el bazo y los predispone a sepsis por neumococo. Para disminuir la mortalidad infantil en estos pacientes es importante la vacunación precoz frente a neumococo y la profilaxis con penicilina.
Anemia hemolítica: estos niños presentan una anemia crónica variable (hemoglobina basal entre 6 y 11g/dl). Pueden presentar crisis aplásicas agudas y graves por infección por parvovirus B19 que precisan transfusión urgente.
Complicaciones vasooclusivas agudas:Crisis de dolor óseo: son la principal causa de ingreso y se deben a infarto óseo. En los niños menores de 2 años afecta a huesos de manos y pies (dactilitis) y en los más mayores a otros huesos. A menudo, precisan ingreso y tratamiento con morfina.
Secuestro esplénico: es una de las complicaciones en los primeros años de vida. Cursa con anemización brusca por secuestro de sangre en el bazo y puede ser mortal. Requiere transfusión urgente.
Síndrome torácico agudo (STA), definido como la aparición de un infiltrado radiológico junto con síntomas respiratorios. Puede deberse a vasooclusión de la circulación pulmonar, neumonía o embolia grasa. En niños es más frecuente la causa infecciosa, principalmente por neumococo y Mycoplasma. Puede empeorar rápidamente, por lo que se debe ingresar al paciente para administrar antibióticos y observación. Puede requerir oxígeno, transfusión y, en los casos más graves, exanguinotransfusión.
Accidente cerebral vascular: tiene lugar hasta en un 10% de los niños (pico de incidencia entre 2 y 6 años). En la infancia suele ser isquémico. El tratamiento consiste en exanguinotransfusión urgente y, para prevenir recurrencias, transfusión crónica. La profilaxis primaria se indica si la ecografía Doppler muestra un flujo cerebral muy aumentado.
Complicaciones crónicas: pueden afectarse distintos órganos como el riñón (microalbuminuria e insuficiencia renal), ojos (glaucoma, desprendimiento de retina), pulmón (hipertensión pulmonar), osteoarticulares (necrosis avascular) y otros.
TratamientoEl tratamiento específico se prescribe a los pacientes con complicaciones importantes. La hidroxiurea aumenta la hemoglobina fetal y disminuye la frecuencia de crisis de dolor y STA. El trasplante de médula ósea se reserva para los pacientes graves que tengan un hermano compatible. La transfusión crónica está indicada como profilaxis primaria y secundaria del ACV y otras complicaciones graves.
El papel del pediatra general es fundamental para poder diagnosticarla de forma precoz, coordinar el seguimiento con otros especialistas, realizar educación sanitaria y consejo genético. Estas medidas son necesarias para reducir la mortalidad infantil de estos niños y mejorar su calidad de vida.
Síndromes drepanocíticos: características clínicas y analíticas
| Síndromes drepanocíticos | Estudio de Hb en < 2 mesesa | Estudio de Hb en > 1 añoa | Hemograma y datos de hemólisis (> 1 año) | VCM | Fenotipo habitualb en un progenitor | Fenotipo habitualb en el otro progenitor | Gravedad |
| SS | FS | SF | Anemia hemolítica | Normal o aumentado | AS | AS | ++++ |
| SC | FSC | SCF | Anemia hemolítica leve o hemólisis compensada | Normal o disminuido | AS | AC | +++ |
| Sβ0 | FS | SF | Anemia hemolítica | Disminuido | AS | Betatalasemia minor (VCM disminuido y HbA2 > 3,5%) | ++++ |
| Sβ+ | FSA | SFA | Anemia hemolítica leve o hemólisis compensada | Normal o disminuido | AS | Betatalasemia minor (VCM disminuido y HbA2 > 3,5%) | ++ |
| Rasgo o portador AS | FAS | AS | No anemia ni hemólisis | Normal | AS | AA o AS | - |
Hb: hemoglobina; VCM: volumen corpuscular medio.
El estado de portador de la anemia falciforme (heterocigoto o AS) se ha asociado a protección frente a la malaria y la prevalencia de esta enfermedad coincide geográficamente con las áreas donde históricamente la incidencia de malaria ha sido más alta7,8. La prevalencia más alta de la mutación se halla en el África subsahariana, pero también afecta al Magreb, Grecia, Arabia Saudí e India9. Debido los movimientos migratorios, su prevalencia es alta en América (afroamericanos) y, más recientemente, en algunos países de Europa10.
DiagnósticoEl diagnóstico se sospecha por morfología eritrocitaria (presencia de hematíes falciformes, véase la figura 1) y se confirma por estudio de hemoglobinas (electroforesis o cromatografía HPLC). Debido a la sustitución de un aminoácido, la HbS tiene una carga y un peso molecular diferentes de la HbA y ello hace que tenga una movilidad distinta y aparezca una banda anómala en la electroforesis o un pico de distinta movilidad por HPLC. En los casos de individuos heterocigotos (AS) mayores de un año, se observa un pico o banda de HbS alrededor del 30%, siempre inferior al 50%, mientras que en los individuos homocigotos SS o en los Sβ, la HbS será la mayoritaria. En los individuos doble heterocigotos SC, la HbS y la HbC constituirán alrededor del 50% cada una. Estas proporciones varían en los primeros meses de vida, en los que predomina la HbF y el porcentaje de HbS será menor (véase la tabla 1).
 de una extensión de sangre periférica en la que se pueden observar los hematíes alargados o falciformes, característicos de esta enfermedad.")
El diagnóstico también puede confirmarse por técnicas moleculares, en las que se detecta el cambio de un nucleótido por otro. El diagnóstico molecular se utiliza para el diagnóstico prenatal.
La ECF debe sospecharse ante todo paciente que presente alguna complicación característica de la enfermedad (anemia hemolítica, dolor óseo o esplenomegalia, entre otras) y sea de grupo étnico de riesgo o proceda de alguna zona geográfica de alta prevalencia. En España, debido a la inmigración, la mayoría de los casos son en individuos de raza negra de origen subsahariano pero también se observa en personas de origen familiar de Magreb y de Latinoamérica2.
Manifestaciones clínicasLa ECF tiene un fenotipo muy variable. Hay pacientes que presentan pocas complicaciones y otros que precisan varios ingresos al año y padecen una enfermedad grave11,12. Las principales manifestaciones clínicas se deben, por un lado, a la hemólisis y, por otro, a los fenómenos vasooclusivos. Estos pacientes presentan complicaciones agudas y otras crónicas por la vasculopatía progresiva que afecta a distintos órganos.
Complicaciones agudas y su tratamientoInfeccionesUna de las principales complicaciones de la drepanocitosis en la edad pediátrica, principalmente en los primeros años de vida, es la sepsis neumocócica. Debido a fenómenos vasooclusivos se producen infartos esplénicos que, ya en los primeros meses de vida, producen una pérdida de función esplénica13. Este hipoesplenismo funcional resulta en una susceptibilidad aumentada a presentar infecciones graves por gérmenes encapsulados, como el neumococo, Haemophilus influenzae y meningococo. Por ello, estos pacientes, en caso de fiebre, deben recibir tratamiento con ceftriaxona, cefotaxima u otro antibiótico que cubra estos gérmenes. Se debe considerar la posibilidad de sepsis neumocócica ante todo paciente con esta enfermedad y fiebre10. En los niños menores de un año con fiebre se recomienda ingreso.
Otra infección que presentan estos enfermos con mayor frecuencia es la osteomielitis. El principal agente causante es la Salmonella, seguido en frecuencia por el S. aureus. El diagnóstico diferencial entre osteomielitis y crisis vasooclusiva ósea a menudo es difícil. Ambos pueden cursar con fiebre, dolor e inflamación local. La gammagrafía ósea no permite distinguir una entidad de otra. La osteomielitis puede sospecharse en los casos de fiebre alta y leucocitosis importante con desviación a la izquierda, y confirmarse por aislarse en cultivo obtenido por punción o biopsia ósea o en hemocultivo. La resonancia magnética puede ser de ayuda en algunos casos. Se debe tener en cuenta que el infarto óseo por crisis vasooclusiva es mucho más frecuente que la osteomielitis y, en los casos que curse con fiebre, se puede iniciar tratamiento para la crisis vasooclusiva y valorar la posibilidad de osteomielitis si la evolución de la misma es tórpida o si se confirma por datos analíticos o microbiológicos14.
Crisis vasooclusivas o de dolorEl dolor es uno de los síntomas principales de la ECF y suele deberse a crisis vasooclusivas óseas. La intensidad y la frecuencia de las crisis de dolor óseo son muy variables entre pacientes. En los casos graves, se manifiestan en forma de dolor intenso y pueden asociarse calor, edema, impotencia funcional y fiebre. A menudo, es necesario tratamiento con morfina. En los niños menores de 2 años estas crisis afectan a los huesos de las manos y los pies (dactilitis o síndrome de mano-pie). En los niños de mayor edad, las crisis dolorosas son más frecuentes y suelen afectar a la columna vertebral, la pelvis y los huesos largos. Estos pacientes pueden también tener crisis de dolor abdominal pero este diagnóstico debe ser uno de exclusión, tras haberse descartado otras causas de dolor abdominal agudo12,14.
Anemia agudaLa hemólisis crónica da lugar a una anemia crónica que puede variar entre pacientes, con hemoglobinas basales entre 6 y 11 g/dl. Los pacientes pueden presentar agudización de su anemia basal. Las principales causas de anemización aguda en estos niños son el secuestro esplénico, las crisis aplásicas y la hiperhemólisis en el contexto de una infección o de una crisis vasooclusiva12,15,16.
Secuestro esplénico: se debe al atrapamiento de drepanocitos en la circulación esplénica que provoca un secuestro sanguíneo en el bazo. Da lugar a un aumento del tamaño del bazo, anemización brusca y, con frecuencia, descenso de la cifra de plaquetas. En las formas graves puede ocasionar shock hipovolémico y ser mortal en menos de una hora, si no se transfunde urgentemente. Es muy importante enseñar a los padres a palpar el bazo y a reconocer los síntomas (astenia marcada, palidez, aumento del tamaño del bazo) para que puedan acudir urgentemente a un hospital en caso de sospecha de esta complicación.
Crisis aplásica: en los niños con ECF, puede tener lugar una eritroblastopenia transitoria, generalmente debida a infección por parvovirus B19. Debido a que estos pacientes requieren de una eritropoyesis aumentada para compensar su hemólisis crónica, se produce una anemia aguda grave reticulocitopénica que suele precisar transfusión10.
Síndrome torácico agudoEl síndrome torácico agudo (STA) es una de las complicaciones más frecuentes de la ECF y uno de los principales motivos de ingreso hospitalario. En ocasiones, se presenta tras una crisis de dolor óseo. Se define por la presencia de clínica respiratoria (tos, dolor torácico y/o dificultad respiratoria) junto con aparición de un infiltrado nuevo en la radiografía de tórax. Puede deberse a infección (neumonía), vasooclusión de la circulación pulmonar y a embolia grasa. A menudo, una se complica con otra y clínicamente es difícil distinguirlas, por lo que se engloba con la denominación de STA. La gravedad de esta complicación es variable pero puede ser mortal y más de un 10% de los casos requieren ventilación mecánica17. El tratamiento consiste en antibióticos que cubran neumococo y Mycoplasma pneumoniae, broncodilatadores y oxígeno. La transfusión sanguínea está indicada en los casos en que haya un descenso importante de la cifra de hemoglobina o cuando el paciente presente un empeoramiento clínico. Los casos más graves pueden requerir ventilación mecánica y exanguinotransfusión.
Accidente cerebrovascularEl accidente cerebrovascular (ACV) es una de las complicaciones más temibles de la ECF, que tiene lugar hasta en un 10% de los niños y un pico de incidencia entre 2 y 6 años. En la infancia suele ser isquémico aunque puede ser hemorrágico. El tratamiento consiste en exanguinotransfusión urgente con el objetivo de alcanzar una hemoglobina S inferior al 30%. Más de la mitad de los niños presentarán un segundo ACV, pero este riesgo puede disminuir a menos de un 10% si se someten a transfusión crónica, cada 3-6 semanas. La profilaxis primaria, también mediante transfusión crónica, se indica en aquellos pacientes en los que por ecografía Doppler se demuestra un flujo cerebral de la arteria cerebral media muy aumentado, ya que presentan un riesgo elevado de presentar un ACV18,19.
PriapismoEl priapismo es otra complicación vasooclusiva de la ECF. Puede ser de corta duración y recurrente. En ocasiones, puede durar más de 2 h y debe tratarse urgentemente para evitar secuelas graves, como la impotencia eréctil. Si no cede con la micción, la hidratación y la analgesia, está indicado aspirar sangre de los cuerpos cavernosos e inyectar un vasoconstrictor local. Para prevenir nuevos episodios se ha descrito la utilización de agonistas adrenérgicos como la seudoefedrina20.
Complicaciones crónicasDebido a la vasculopatía progresiva en distintos órganos, estos pacientes presentan complicaciones crónicas que ya pueden empezar en la edad pediátrica. Entre ellas destacan por su frecuencia y gravedad las complicaciones renales (microalbuminuria, enuresis, incapacidad de concentrar la orina e insuficiencia renal)21, la hipertensión pulmonar, en especial en aquellos pacientes con mayor hemólisis22,23, la osteonecrosis avascular, el deterioro cognitivo por infartos «silentes»24,25 y las alteraciones oculares (desprendimiento de retina, glaucoma postraumático)26. Todo ello requiere de un seguimiento en una unidad especializada en estos pacientes, multidisciplinar, con especialistas pediátricos que conozcan esta enfermedad.
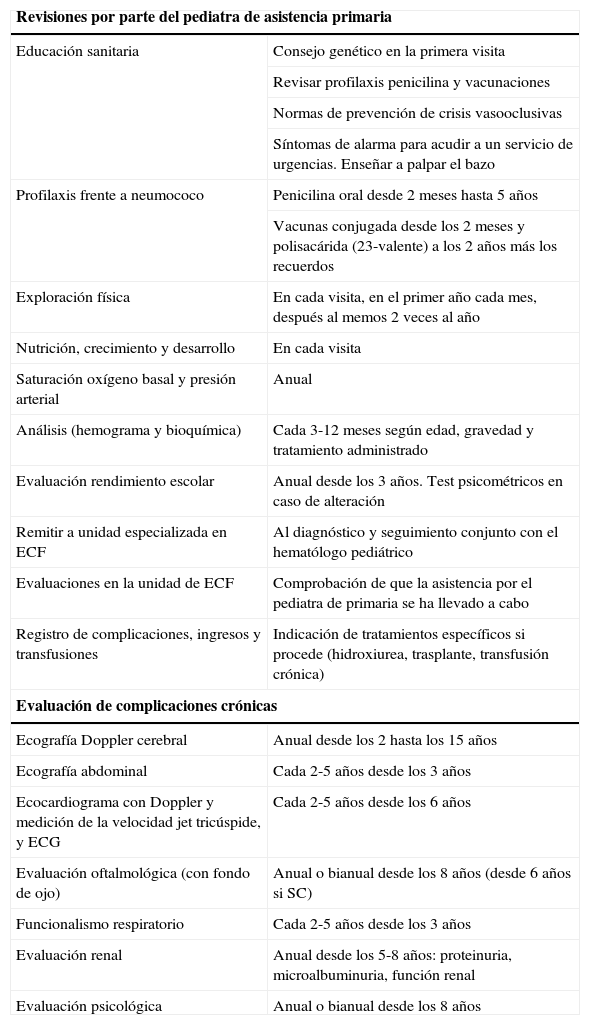
Manejo generalPrevención de las infeccionesUna de las medidas que han demostrado reducir de forma significativa la mortalidad infantil en estos pacientes ha sido la profilaxis de la infección neumocócica27. Se debe iniciar a los 2 meses de edad o en cuanto se conozca el diagnóstico de ECF y consiste en la administración de penicilina por vía oral diaria (al menos hasta los 5 años de vida) y la vacunación precoz frente a neumococo. La vacuna conjugada puede iniciarse ya a los 2 meses de edad y la vacuna polisacárida (23-valente) a partir de los 2 años (véase la tabla 2).
Seguimiento recomendado en los niños con ECFa
| Revisiones por parte del pediatra de asistencia primaria | |
| Educación sanitaria | Consejo genético en la primera visita |
| Revisar profilaxis penicilina y vacunaciones | |
| Normas de prevención de crisis vasooclusivas | |
| Síntomas de alarma para acudir a un servicio de urgencias. Enseñar a palpar el bazo | |
| Profilaxis frente a neumococo | Penicilina oral desde 2 meses hasta 5 años |
| Vacunas conjugada desde los 2 meses y polisacárida (23-valente) a los 2 años más los recuerdos | |
| Exploración física | En cada visita, en el primer año cada mes, después al memos 2 veces al año |
| Nutrición, crecimiento y desarrollo | En cada visita |
| Saturación oxígeno basal y presión arterial | Anual |
| Análisis (hemograma y bioquímica) | Cada 3-12 meses según edad, gravedad y tratamiento administrado |
| Evaluación rendimiento escolar | Anual desde los 3 años. Test psicométricos en caso de alteración |
| Remitir a unidad especializada en ECF | Al diagnóstico y seguimiento conjunto con el hematólogo pediátrico |
| Evaluaciones en la unidad de ECF | Comprobación de que la asistencia por el pediatra de primaria se ha llevado a cabo |
| Registro de complicaciones, ingresos y transfusiones | Indicación de tratamientos específicos si procede (hidroxiurea, trasplante, transfusión crónica) |
| Evaluación de complicaciones crónicas | |
| Ecografía Doppler cerebral | Anual desde los 2 hasta los 15 años |
| Ecografía abdominal | Cada 2-5 años desde los 3 años |
| Ecocardiograma con Doppler y medición de la velocidad jet tricúspide, y ECG | Cada 2-5 años desde los 6 años |
| Evaluación oftalmológica (con fondo de ojo) | Anual o bianual desde los 8 años (desde 6 años si SC) |
| Funcionalismo respiratorio | Cada 2-5 años desde los 3 años |
| Evaluación renal | Anual desde los 5-8 años: proteinuria, microalbuminuria, función renal |
| Evaluación psicológica | Anual o bianual desde los 8 años |
ECF: enfermedad de células falciformes.
El seguimiento de estos pacientes por parte de unidades especializadas en ECF ha mejorado significativamente su pronóstico28,29. Es muy importante un abordaje multidisciplinar y coordinado para el correcto manejo de estos niños. El pediatra de atención primaria desempeña un papel central en su seguimiento. Con frecuencia, será a quien se dirigirán ante una primera complicación de su enfermedad todavía no diagnosticada y quien realizará la derivación a un servicio de urgencias en las complicaciones agudas que lo requieran. También es muy importante que vacune e inicie la profilaxis antibiótica en cuanto tenga el diagnóstico. Podrá colaborar con el especialista en pediatría hematológica en la realización de los controles clínicos y las exploraciones complementarias recomendadas para prevenir y tratar las complicaciones crónicas asociadas a esta enfermedad14 (véase la tabla 2). En este sentido, la Sociedad Española de Hemato-oncología Pediátrica ha realizado una guía clínica de ECF30.
Educación y soporte psicológicoLa ECF en nuestro país afecta, principalmente, a la población inmigrante quien, a menudo, presenta problemas económicos y sociales que se añaden a los ya inherentes de una enfermedad crónica. La educación sanitaria y el soporte psicológico en estos pacientes son, por ello, muy importantes para la prevención y el tratamiento precoz de muchas de las complicaciones. Se debe educar a los pacientes y a sus familias a evitar los factores que puedan precipitar una crisis vasooclusiva, tales como la exposición al frío, la deshidratación y la fiebre. También deben aprender cómo tratar las crisis de dolor leve con aumento de la ingesta de agua, reposo, analgésicos como paracetamol e ibuprofeno, u opiáceos suaves como la codeína. Se debe enseñar a reconocer los signos de alarma que requieran de la atención inmediata en un servicio de urgencias, tales como palidez, astenia, fiebre alta, dificultad respiratoria o aparición de una focalidad neurológica. Por otro lado, el soporte psicológico al paciente y a los familiares es fundamental. Se trata de una enfermedad crónica, con crisis de dolor agudo y a veces dolor crónico, ingresos hospitalarios y complicaciones graves que pueden ser mortales. Todo ello facilita el ausentismo escolar, que en ocasiones se añade a alteraciones cognitivas por infartos cerebrales «silentes» y a necesidad de refuerzo escolar. La adolescencia es una etapa especialmente difícil en estos pacientes crónicos. Con frecuencia, presentan un retraso puberal y es conveniente informarles que, por lo general, alcanzarán un desarrollo sexual y una altura normales en la edad adulta. Otro aspecto importante es la preparación para la transición a la atención en unidades de adultos, ya que se ha descrito una elevada mortalidad en estos pacientes en el paso de la atención sanitaria pediátrica a la de adultos31.
Cribado neonatalPara disminuir la mortalidad infantil de la ECF resulta primordial establecer su diagnóstico lo antes posible. Así pueden tomarse las medidas preventivas de infección neumocócica (vacunación y penicilina) y la educación sanitaria que permita el reconocimiento y el tratamiento precoz de las complicaciones de esta enfermedad. El cribado neonatal mediante estudio de hemoglobinas en sangre de talón es una medida que ha demostrado su eficacia en reducir la mortalidad de estos pacientes32,33. En nuestro país, el cribado neonatal universal se realiza desde 2003 en la Comunidad Autónoma de Madrid34,35 y recientemente en otras comunidades autónomas; sin embargo, en la mayoría del territorio español todavía no se realiza este cribado pese a cumplir los criterios necesarios para su inclusión. En el caso de Cataluña y en Baleares, se han hecho estudios pilotos que estiman una prevalencia de ECF de alrededor de uno por 5.000 neonatos, similar a la incidencia de fenilcetonuria36–39.
Consejo genético y diagnóstico prenatalLa ECF tiene una herencia autosómica recesiva. En el momento en que se diagnostica a un niño, es esencial la evaluación precoz de los hermanos, por si estuvieran también afectados, y de los padres, para poder ofrecerles consejo genético y tratar de evitar nuevos casos. Es importante recordar que, en nuestro medio es frecuente la betatalasemia minor y que, si se hereda por parte de un progenitor una betatalasemia minor y el otro progenitor es portador heterocigoto de HbS, los hijos pueden tener una ECF (doble heterocigota Sβ).
En el caso de que 2 portadores deseen tener más hijos, se debe informar de la posibilidad de un diagnóstico prenatal.
Prevención de las complicaciones postoperatoriasLos pacientes con ECF tienen mayor riesgo de presentar crisis vasooclusivas, como el síndrome torácico agudo, tras una intervención quirúrgica. Ello se debe a la inmovilización y las posibles atelectasias, al frío, a la deshidratación tras el ayuno, a la hipoxemia durante la anestesia y a otros factores de estrés que pueden desencadenar una crisis. Para intentar prevenir estas complicaciones postoperatorias, se deben tomar unas medidas perioperatorias, tales como la hidratación adecuada la noche antes de la intervención, transfusión sanguínea previa a la cirugía mayor, sin exceder una hemoglobina de 10g/dl tras la transfusión, que podría aumentar la viscosidad sanguínea. Durante la intervención quirúrgica, es importante asegurar una buena oxigenación y evitar el frío. En el postoperatorio se debe mantener una correcta oxigenación, un control adecuado del dolor, promover la deambulación precoz y realizar fisioterapia respiratoria con el fin de evitar atelectasias que podrían precipitar crisis vasooclusivas40,41.
TratamientoLa gran variabilidad fenotípica de estos pacientes hace necesario adecuar el tratamiento al curso clínico que presente el paciente. El tratamiento de las complicaciones agudas se ha descrito con anterioridad. A continuación, se describe el tratamiento de los pacientes que presentan un curso moderado a grave de la enfermedad (tabla 3).
Indicaciones de tratamientos específicos en ECF.
| Hidroxiurea |
| Eficacia demostrada |
| Crisis de dolor vasooclusivo grave recurrente (más de 3 ingresos por año) |
| STA recurrente (3 en los últimos 2 años o uno grave) |
| Cualquier combinación de 3 o más episodios de crisis de dolor o STA/año |
| Eficacia probable |
| Anemia basal grave |
| Prevención de accidente vascular cerebral |
| Pacientes con otras complicaciones graves de la enfermedad |
| Prevención de lesiones crónicas en órganos |
| Disminución de la mortalidad |
| Transfusión o exanguinotransfusión crónica (objetivo mantener HbS < 30%) |
| Eficacia demostrada |
| Profilaxis primaria y secundaria de accidente vascular cerebral |
| Eficacia probable o posible |
| Prevención secundaria de secuestro esplénicoa |
| Prevención de crisis recurrentes de dolor graves de repetición que no han respondido a tratamiento con hidroxiurea |
| Prevención de STA de repetición que no han respondido a tratamiento con hidroxiurea |
| Gestación de alto riesgo |
| Insuficiencia renal crónica |
| Hipertensión pulmonar |
| Trasplante de progenitores hematopoyéticosb |
| Criterios de inclusión |
| Pacientes con ECF (SS, SC, S-betatalasemia) de edad < 16 años con hermano HLA idéntico sano o portador |
| Enfermedad grave que haya presentado una o más de las siguientes complicaciones |
| ACV |
| Síndrome torácico agudo grave o de repeticiónc |
| Crisis vasooclusivas dolorosas recurrentesc |
| Disfunción neuropsicológica con RM craneal anormal |
| Enfermedad pulmonar falciforme en estadios I–II |
| Nefropatía falciforme (filtrado glomerular 30–50 del valor normal) |
| Aloinmunización múltiple en un paciente que está en régimen de transfusión crónica |
| Otras manifestaciones de ECF grave (retinopatía proliferativa bilateral con afectación visual y otras) |
| Criterios de exclusión |
| Enfermedad con afectación neurocognitiva, pulmonar, hepática o renal graves o estado general muy afectado |
ACV: accidente cerebrovascular; ECF: enfermedad de células falciformes; RM: resonancia magnética; STA: síndrome torácico agudo.
La hidroxiurea es un citostático que se utiliza en los pacientes con formas graves de ECF, que induce la formación de hemoglobina fetal y, con ello, reduce la falciformación. Su efecto beneficioso se produce también por otros mecanismos, como la reducción de la cifra de leucocitos, la disminución de la interacción de las células sanguíneas con el endotelio y el aumento de producción de NO. Con este tratamiento se consigue un aumento de la hemoglobina basal y de la fetal, y se disminuyen las crisis de dolor y de síndrome torácico agudo, con la consecuente reducción de ingresos y de transfusiones42–45. Además, en algunos estudios en adultos46 y, recientemente, en un estudio pediátrico47, se ha observado una disminución de la mortalidad de aquellos pacientes que reciben este tratamiento. Con un seguimiento de más de 25 años en adultos y de más de 15 en niños, se ha podido demostrar un beneficio a largo plazo y un perfil de seguridad aceptable. No se han observado carcinogénesis ni alteraciones en el crecimiento. Sin embargo, se requiere monitorización con hemogramas periódicos para ajustar la dosis y evitar citopenias. Además, debe informarse de su potencial teratogénico y de la posibilidad de azoospermia45.
Transfusión sanguínea crónica y quelaciónLa transfusión en la ECF, además de corregir la anemia, tiene como beneficio adicional la reducción del porcentaje de HbS. La transfusión sanguínea simple está indicada en algunas complicaciones agudas. La transfusión crónica, cada 3-6 semanas, se indica como profilaxis primaria o secundaria de los ACV y en algunas complicaciones crónicas de la ECF. En algunas situaciones puede requerirse la exanguinotransfusión, cuando la hemoglobina basal es alta o cuando se requiere de un descenso rápido del porcentaje de HbS, como en los ACV. La exanguinotransfusión puede realizarse de forma manual o automatizada, mediante eritroaféresis. En aquellos pacientes que requieren transfusión crónica es preciso asociar tratamiento quelante del hierro para evitar la sobrecarga férrica cardiaca y hepática, y otras manifestaciones de la hemosiderosis secundaria48.
Trasplante de médula óseaEl trasplante de progenitores hematopoyéticos obtenidos de médula ósea o de cordón umbilical está indicado en los pacientes con enfermedad más grave, como aquellos que están en régimen de transfusión crónica. Es el único tratamiento curativo de la ECF; sin embargo, solo algunos de los pacientes que tienen indicación tienen un hermano no afectado y HLA compatible. No está indicado en todos los pacientes con hermano compatible debido a la morbimortalidad asociada al trasplante. Según la edad y la morbilidad de los pacientes, el trasplante en estos niños tienen una supervivencia global entre el 92 y el 94%, una tasa de recaídas o fallo de implante de alrededor del 10%. A la hora de decidir este tratamiento, se deben evaluar, conjuntamente con el paciente y la familia, no solo los riesgos de mortalidad y de recaída, sino también la morbilidad que puede tener en algunos casos, como la infertilidad y la enfermedad del injerto contra el huésped crónica49.
Perspectivas futurasComo perspectivas de futuro en la ECF, se están evaluando nuevos tratamientos inductores de la producción de hemoglobina fetal, sildenafilo y otros fármacos que aumenten la disponibilidad de NO en la hipertensión pulmonar, nuevas modalidades de trasplante de progenitores hematopoyéticos y la terapia génica50.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.