


La fibrosis quística (FQ) es la enfermedad genética grave, de herencia autosómica recesiva, más frecuente en las poblaciones de origen caucásico; su incidencia se sitúa entre 1/2.500 y 1/6.000 recién nacidos vivos. El gen responsable codifica para una proteína, regulador de la conductancia transmembrana de la fibrosis quística (CFTR), que se comporta como un canal para el cloro, situado en la membrana apical del epitelio secretor de las mucosas del aparato respiratorio, digestivo y reproductor, así como en las glándulas del sudor y la saliva. Las manifestaciones clínicas más frecuentes son la enfermedad pulmonar obstructiva crónica, la insuficiencia pancreática, la azoospermia obstructiva y las concentraciones elevadas de cloro y sodio en el sudor, que pueden llevar a situaciones de deshidratación o golpe de calor. Casi todos los pacientes presentan clínica respiratoria y su gravedad determina el pronóstico vital, mientras que la insuficiencia pancreática aparece en el 85–90% de los casos1.
Puntos clave
- •
El cribado neonatal de la fibrosis quística permite realizar el diagnóstico, antes de que se manifieste la clínica, e iniciar un tratamiento precoz.
- •
Los protocolos habitualmente empleados incluyen la determinación de tripsina inmunorreactiva (TIR) entre el 3.er y 5.° día de vida. Si está elevada, se realiza estudio genético (protocolo TIR/ADN) o se repite la determinación de TIR entre los 25–40 días de vida (protocolo TIR/TIR).
- •
La positividad del test del sudor, realizado en una unidad de referencia, confirma el diagnóstico.
- •
El seguimiento se hará en una unidad de referencia y conforme a las guías internacionales publicadas. Los lactantes con test del sudor dudosos también serán objeto de seguimiento y ulteriores pruebas diagnósticas.
- •
Los lactantes asintomáticos pueden tener una función pulmonar y una tomografía computarizada torácica anómalas.
- •
La implantación del cribado neonatal ha mejorado los resultados clínicos y ha disminuido la incidencia de la enfermedad.
La implantación de un programa de cribado neonatal, para una determinada enfermedad, tiene como objetivo realizar un diagnóstico temprano de la misma, incluso antes de que aparezcan las primeras manifestaciones clínicas. Con ello se evitan diagnósticos tardíos o erróneos, se refuerza la confianza de los padres, se inicia precozmente el tratamiento adecuado y se proporciona consejo genético a la familia.
Para que esté indicado el cribado neonatal de una determinada enfermedad, se debe cumplir una serie de criterios: a) que la enfermedad tenga una incidencia importante; b) que se disponga de un método de cribado simple y fácil de realizar; c) que este tenga un alto grado de sensibilidad y especificidad; d) que la relación coste-beneficio sea buena, y e) que la instauración precoz de un tratamiento modifique, de forma favorable, el curso de la enfermedad. Estos criterios se cumplen, en el caso de la FQ. Por ello, a finales de la década de los setenta, se iniciaron programas de cribado neonatal en algunos países. Francia fue el primero que, en 2002, adoptó el cribado en todo el territorio nacional. Al año siguiente, Australia, que había iniciado su programa de cribado en 1981, lo extendió a todo el país. En Estados Unidos, que fue pionero en este campo, se fue incorporando paulatinamente en los distintos estados y a finales del 2010 se realizaba en la práctica totalidad de los mismos.
En España, el cribado neonatal de la FQ se inició a finales de los noventa, siendo Cataluña y Castilla y León las primeras comunidades autónomas en llevarlo a cabo. En la actualidad, se practica en 11 comunidades. Durante este tiempo, se ha estudiado a más de 1,5 millón de recién nacidos y se han realizado más de 300 diagnósticos2.
Hasta el momento, se han publicado diversos estudios que reflejan los resultados del seguimiento realizado a estos pacientes y que han resultado muy interesantes. En primer lugar, se ha podido disponer de cifras reales sobre la incidencia de la enfermedad en cada país. Esta difiere ampliamente. Así, en Europa, encontramos un caso por cada 25.000 recién nacidos en Finlandia, mientras que en Eslovenia tienen un caso por cada 1.800. La mayoría de los países europeos presentan una incidencia intermedia: 1:2.381 en Reino Unido, 1:3.300 en Alemania, 1: 4.238 en Italia y 1:4.348 en Francia3. En poblaciones de origen no europeo, la incidencia es más baja, siendo extremo el caso de Japón, donde la FQ es excepcional. En España, los registros de las distintas comunidades proporcionan cifras que oscilan entre uno por cada 6.496 recién nacidos en Cataluña y uno por cada 4.500 en Castilla y León4.
El cribado neonatal también ha permitido conocer mejor la historia natural de la enfermedad.
Por ejemplo, se ha podido comprobar que a los 3 meses de vida ya se pueden detectar anomalías en las pruebas de función respiratoria. Así se ha visto que estos niños presentan un aumento de la capacidad residual funcional, mayor índice de aclaramiento pulmonar y flujos espiratorios más bajos5. Otros autores han detectado, mediante lavado broncoalveolar (LBA), Pseudomonas aeruginosa [P. aeruginosa] en el 15% de los niños, correlacionándose su presencia con el aumento de neutrófilos y de IL-86. Incluso en niños sin síntomas respiratorios, se ha podido observar, en el LBA, la presencia de bacterias en el 21%, así como aumento de neutrófilos, de IL-8 y de elastasa neutrofílica. Además, mediante tomografía computarizada (TAC) del tórax se han detectado anomalías radiológicas en el 80% de los casos7. En otros estudios se ha encontrado, al año de vida, la presencia de bronquiectasias en el 27% y atrapamiento aéreo en el 49% de los lactantes8.
Por otra parte, se han publicado distintos artículos que encuentran que la evolución clínica es mejor en los pacientes con FQ diagnosticados por cribado. Aunque se han realizado pocos ensayos clínicos aleatorizados, parece que estos niños tienen un estado nutricional mejor que los diagnosticados por la clínica9. Los primeros trabajos observacionales ya apuntaban en esta dirección10,11. Además, se ha visto que estos pacientes tienen menor tasa de colonización por P. aeruginosa y mejor función pulmonar12,13. Por lo tanto, no es de extrañar que los niños FQ, diagnosticados por cribado, tengan mayor supervivencia14,15, incluso con menor gasto sanitario, que los diagnosticados más tardíamente16.
Por todo lo tanto, en el momento actual, el cribado neonatal de FQ está plenamente justificado.
Técnicas y protocolos empleadosDurante los años sesenta, se empleó la detección de albúmina en el meconio como método de cribado. A finales de los setenta, se observó que los neonatos con FQ presentaban aumento de la tripsina inmunorreactiva (TIR) en sangre17. Al tratarse de una técnica fácil de realizar, fiable y con alta sensibilidad, se propuso su determinación, en la muestra de sangre de talón, para realizar el cribado neonatal de la enfermedad. Hay que señalar que los pacientes FQ con íleo meconial pueden tener niveles de TIR normales18. Por otra parte, factores como la prematuridad o la pertenencia a la raza negra pueden elevar los niveles de TIR en recién nacidos sanos19,20. Lo mismo sucede en portadores de alguna mutación de FQ21.
El descubrimiento del gen de la FQ en 198922, en el brazo largo del cromosoma 7 (7q31.2), ha proporcionado una herramienta de primer orden para realizar el diagnóstico, Hernández como es el estudio de las mutaciones relacionadas con la enfermedad23, de las que hasta el momento hay más de 1.800 descritas. El procedimiento resulta más caro que la determinación de la TIR, por lo que se recomienda realizarlo solo si esta última está elevada. Debe utilizarse un panel de estudio que incluya un número suficiente y representativo de las mutaciones más frecuentes en la población estudiada24.
Menos extendida está la determinación, en la gota de sangre del talón, de la proteína asociada a la pancreatitis (pancreatitis associated protein). Se ha preconizado su inclusión, junto con la TIR, en los protocolos de cribado de FQ, evitando así la realización del estudio genético. Esto abarataría el precio del cribado, aunque disminuiría algo la especificidad25,26.
Dependiendo de la variabilidad genética de la población y de su capacidad económica, se han diseñado diversos programas de cribado neonatal. En todos los casos, se parte de la determinación de TIR en la muestra de sangre obtenida, por punción del talón del recién nacido, entre el 3.er y 5.° día de vida, que impregna la tarjeta correspondiente. Se recomienda realizar determinaciones seriadas en la población de estudio para situar el punto de corte de tal manera que se minimice el riesgo de falsos negativos. Si esta primera determinación de TIR resulta normal, el resultado es negativo y se comunica a la familia. En caso contrario, se sigue con alguno de los estos protocolos27:
- 1.
Tripsina inmunorreactiva/tripsina inmunorreactiva
Se realiza una segunda determinación de TIR entre los días 25 y 40 de vida. Si el resultado está dentro de los límites normales, el cribado se considera negativo. Si la TIR continúa elevada, se remite al niño a una unidad de referencia, para su valoración y la realización del test del sudor. Esta estrategia es más barata y no identifica portadores, aunque teóricamente podrían perderse sujetos que no acudieran a realizarse la segunda determinación.
- 2.
Tripsina inmunorreactiva/ADN
En la misma muestra de sangre de talón, de aquellos niños que hayan presentado una primera TIR elevada, puede hacerse el estudio genético de mutaciones de la FQ. Con esta estrategia, aumentan la sensibilidad y especificidad. Por la frecuencia de la mutación F508del, en el norte de Europa, el panel de mutaciones empleado es inferior al de la zona mediterránea. Así, en España, esta mutación solo se encuentra en el 50% de los alelos estudiados. Además, existe una amplia variedad de mutaciones, producto de las corrientes migratorias históricas28,29, que obliga a ampliar el panel de estudio. Si en el estudio genético se encuentra alguna mutación, el niño es remitido a una unidad de referencia. Si el estudio es negativo, se realiza la segunda determinación de TIR y, si continúa elevada, también se remite a la unidad de referencia.
La decisión de adoptar un protocolo u otro estará en manos de las autoridades sanitarias de cada comunidad. El protocolo TIR/ADN tiene la ventaja de realizar el diagnóstico más rápidamente y no precisar una segunda muestra de sangre en muchos casos, aunque resulta más caro que el protocolo TIR/TIR30.
La unidad de referencia debe ser una unidad multidisciplinaria, que cuente con personal experto en el diagnóstico y el seguimiento de la FQ. En ella se realizará el diagnostico definitivo de los niños derivados desde el programa de cribado neonatal. Para ello se practica un test del sudor, que se considera positivo si la concentración de cloro es igual o superior a 60mmol/l. Dos pruebas positivas confirman el diagnóstico de FQ. Los valores situados entre 30 y 59mmol/l se consideran dudosos, siendo necesario practicar otras pruebas complementarias y realizar un seguimiento clínico31. Hay que tener en cuenta que en los lactantes más pequeños, así como en los prematuros, la producción y la recogida del sudor pueden ser dificultosas. Por ello se recomienda realizar la prueba a partir de la 2.a–3.a semana de vida y cuando el niño pesa más de 3kg32.
Con los resultados del test del sudor y el estudio genético, se plantean las siguientes situaciones:
- –
Falso positivo: test del sudor normal y estudio genético negativo.
- –
Diagnóstico de FQ: test del sudor positivo y/o estudio genético con 2 mutaciones.
- –
Portador: test del sudor normal y estudio genético con una mutación.
- –
No concluyente: test del sudor con valores dudosos y detección de una o ninguna mutación en el estudio genético.
En este último grupo es aconsejable realizar un estudio genético ampliado y seguir al paciente, con repetición periódica de test del sudor33,34. Tanto a los padres de los niños enfermos, como a los de los portadores, se les ofrece consejo genético. Esto probablemente ha contribuido al descenso de casos de FQ observado en países donde el cribado viene realizándose desde hace tiempo35.
Protocolo de seguimientoLa Unidad Multidisciplinaria de FQ debe contar con protocolos de seguimiento para los lactantes diagnosticados por cribado neonatal. Como primer paso, se debe dar información a los padres sobre la enfermedad, el tratamiento y el pronóstico, proporcionando apoyo psicológico y despejando dudas36. Es aconsejable realizar un test del sudor a los hermanos.
El programa de seguimiento incluye visitas periódicas y la realización de pruebas complementarias. Al principio, las visitas suelen realizarse mensualmente y a partir del 6.°–8.° mes, cada 2–3 meses, dependiendo de la evolución. En cada visita, se evaluará clínicamente al niño desde el punto de vista nutricional y para detectar precozmente las manifestaciones digestivas o respiratorias de la enfermedad37. La realización de pruebas complementarias dependerá del estado del niño y del curso de la enfermedad. En general, en la primera visita se realiza analítica sanguínea y determinación de elastasa fecal para descartar la existencia de insuficiencia pancreática. En posteriores visitas, se seguirá el protocolo habitual de los pacientes FQ38, ajustado a las necesidades de cada niño.
No hay consenso sobre el momento de realizar la primera radiografía de tórax y/o TAC torácica en un lactante asintomático, y varía, según las distintas unidades, desde momento del diagnóstico hasta los 12–24 meses. Existen menos dudas de si el paciente presenta reagudizaciones respiratorias. A la hora de practicar una TAC torácica, se recomienda emplear dosis bajas de radiación, hacerlo bajo sedación y realizar maniobras de inspiración y espiración para valorar la pequeña vía aérea39,40.
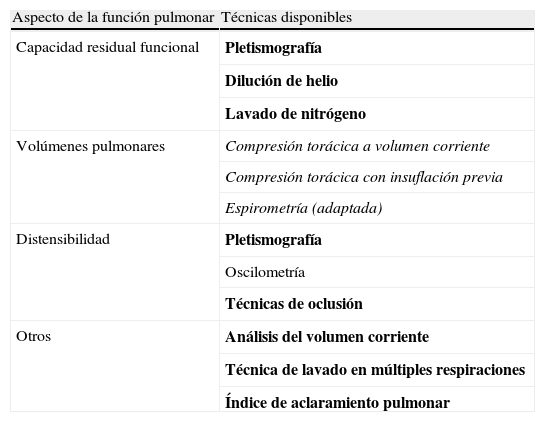
Para realizar pruebas de función respiratoria en lactantes, se precisa disponer de equipos costosos y personal adiestrado, por lo que su práctica en estos niños suele tener fines de investigación. A partir de los 3–4 años, el niño suele colaborar lo suficiente para realizar espirometrías. En la tabla 1 se enumeran las técnicas disponibles para realizar las pruebas de función respiratoria en lactantes y en preescolares. Tampoco se suele realizar LBA de rutina, aunque en casos de reagudización respiratoria grave, con mala respuesta al tratamiento, podría estar indicado para precisar el diagnóstico microbiológico.
Pruebas de función respiratoria en niños pequeños.
| Aspecto de la función pulmonar | Técnicas disponibles |
| Capacidad residual funcional | Pletismografía |
| Dilución de helio | |
| Lavado de nitrógeno | |
| Volúmenes pulmonares | Compresión torácica a volumen corriente |
| Compresión torácica con insuflación previa | |
| Espirometría (adaptada) | |
| Distensibilidad | Pletismografía |
| Oscilometría | |
| Técnicas de oclusión | |
| Otros | Análisis del volumen corriente |
| Técnica de lavado en múltiples respiraciones | |
| Índice de aclaramiento pulmonar |
Técnicas disponibles en lactantes (cursivas), preescolares (normal) y en ambos (negrita).
El tratamiento se realizará con enzimas pancreáticas, cuando exista insuficiencia pancreática, administrándose también vitaminas liposolubles y aportes extra de sales, especialmente en épocas de calor. En cuanto a la dieta, se aconseja la lactancia materna, introduciendo la alimentación complementaria según edad y realizando ajustes periódicos de enzimas y vitaminas.
Desde el punto de vista respiratorio, se recomienda iniciar la práctica de fisioterapia respiratoria y la administración de las vacunas correspondientes, según el calendario vacunal, sin olvidar la vacuna antineumocócica. También se recomienda la vacuna antigripal a los padres y familiares y a los lactantes a partir de los 6 meses. Aunque no existe una opinión unánime al respecto, podría considerarse la profilaxis frente al virus sincitial respiratorio con palivizumab durante la época epidémica41. La administración de medicación inhalada (broncodilatadores, corticoides) suele reservarse a los lactantes con sibilancias de repetición, aunque no existen trabajos que avalen su eficacia. Respecto a los fármacos que mejoran el aclaramiento mucociliar, se ha visto que tanto el suero salino hipertónico42 como la dornasa alfa43 son bien tolerados por los niños más pequeños, pero no se ha podido demostrar su eficacia a esta edad44.
Para el control de la infección respiratoria en cada visita se obtiene una muestra de exudado faríngeo. En caso de reagudización respiratoria, se iniciará tratamiento antibiótico según el germen aislado en el último cultivo. Al principio, los niños se suelen colonizar por Staphylococcus aureus (S. aureus), estando indicada la administración de antibióticos en caso de exacerbación respiratoria. Las guías de tratamiento británicas preconizan el uso de antibióticos por vía oral de forma profiláctica, administrando flucloxacilina desde el diagnóstico hasta los 3 años, pero no todos los grupos están de acuerdo con esta práctica45. Otros autores, empleando cefalexina, observaron que efectivamente se retrasaba la colonización por S. aureus, pero aumentaba el número de pacientes colonizados por P. aeruginosa y no se observaba beneficio clínico46. Otra alternativa sería la de administrar antibióticos frente a S. aureus durante 2–4 semanas, si este germen se aísla en el cultivo de exudado faríngeo. Sí hay acuerdo en tratar el primer aislamiento de P. aeruginosa, habiéndose publicado diferentes protocolos terapéuticos. En el de la Sociedad Española de Neumología Pediátrica, se preconiza la administración de ciprofloxacino por vía oral durante 3 semanas y colistina o tobramicina inhaladas de forma continua. Si al cabo de un mes de tratamiento el cultivo sigue siendo positivo, se continúa con la terapia inhalada y se administra un nuevo ciclo de ciprofloxacino por vía oral. Si no se consigue negativizar el cultivo, se puede administrar un nuevo ciclo de antibióticos anti-Pseudomonas, por vía intravenosa, y se mantiene el tratamiento inhalado durante 6–12 meses hasta obtener 3 cultivos negativos, en muestras tomadas con 1–2 meses de intervalo47. Si al cabo de este tiempo no se ha negativizado el cultivo, se considera que la colonización se ha cronificado. Otros autores han conseguido buenas tasas de erradicación empleando solo tobramicina inhalada durante 28 días48. Con la política de erradicación, se eleva la edad en la que se produce la colonización definitiva y disminuye la prevalencia de la infección crónica49. Se han realizado ensayos clínicos, administrando antibióticos anti-Pseudomonas de forma profiláctica, pero por ahora no han demostrado su eficacia50.
Como resumen, cabe resaltar el avance que ha supuesto la implantación del cribado neonatal de la FQ en las distintas comunidades. A las ventajas innegables de realizar un diagnóstico precoz de la enfermedad e iniciar un tratamiento adecuado, incluso antes de que la clínica sea manifiesta, se une la posibilidad de estudiar mejor la enfermedad desde el principio. En este sentido, se han diseñado distintas líneas de investigación que se podrían resumir en los siguientes puntos:
- –
Profundizar en el conocimiento de los mecanismos patogénicos de la FQ en sus fases iniciales.
- –
Conocer mejor la historia natural de la enfermedad pulmonar y detectar cambios precoces producidos por los mecanismos de la inflamación y de reparación, así como la
- –
influencia que en estos procesos puedan ejercer los distintos gérmenes que habitualmente colonizan la vía aérea de estos pacientes.
- –
Encontrar biomarcadores que señalen cambios patológicos y sirvan para evaluar los resultados clínicos obtenidos tras el tratamiento oportuno.
- –
Evaluar la respuesta al tratamiento dependiendo del genotipo del paciente, para poder diseñar tratamientos individualizados y más eficaces.
Por todo ello, es deseable que el cribado neonatal se generalice y que se potencie la investigación en este campo.