






Puntos clave
- •
La principal causa de la coagulación intravascular diseminada (CID) en niños es la sepsis.
- •
No existe una prueba diagnóstica de CID; el diagnóstico se basa en la presunción clínica junto con determinadas pruebas de laboratorio.
- •
El principal tratamiento de la CID es el tratamiento de la enfermedad que la ha desencadenado.
- •
La alteración de la coagulación en la enfermedad hepática es multifactorial, con alteraciones que predisponen al sangrado y otras que predisponen a la trombosis.
- •
El hallazgo más frecuente en la coagulopatía de la enfermedad hepática es el alargamiento del tiempo de protrombina y el factor VII es el primero que disminuye.
- •
El tratamiento de la coagulopatía en la enfermedad hepática está indicado en situaciones de sangrado y previo a la realización de procedimientos invasivos.
Las alteraciones de la coagulación son afecciones frecuentes en el paciente pediátrico crítico, estando implicadas en la fisiopatología de las mismas la respuesta inflamatoria, las alteraciones de la hemostasia y la disfunción endotelial. Constituyen una complicación frecuente en estos pacientes y asocian una elevada morbimortalidad. La coagulopatía asociada a sangrado es una entidad relativamente frecuente en el paciente pediátrico crítico y suele ocurrir en el contexto de enfermedad sistémica. Son múltiples las causas de coagulopatías con sangrado en la Unidad de Cuidados Intensivos Pediátricos (tablas 1 y 2), siendo el objeto de la presente actualización la coagulación intravascular diseminada (CID) y la coagulopatía asociada a enfermedad hepática por su gravedad y relevancia clínica1.
Lectura rápida
Las alteraciones de la coagulación son afecciones frecuentes en el paciente pediátrico crítico, estando implicadas en la fisiopatología de las mismas la respuesta inflamatoria, las alteraciones de la hemostasia y la disfunción endotelial. Constituyen una complicación frecuente en estos pacientes y asocian una elevada morbimortalidad.
La coagulación intravascular diseminada (CID) es un trastorno que puede aparecer en diferentes enfermedades. La sepsis es la principal causa de CID en niños.
La CID se produce por una activación excesiva e incontrolada de la coagulación, que conduce a la aparición generalizada de trombosis intravascular. A su vez, el consumo de factores va a conducir a un descenso de los mismos, apareciendo fenómenos hemorrágicos.
La activación del factor tisular, la disminución de la actividad de los anticoagulantes y la inactivación de la fibrinólisis son los principales factores que intervienen en la aparición del estado procoagulante de la CID.
Las manifestaciones clínicas de la CID van a ser muy variables, dependiendo de la severidad del cuadro y de la velocidad de instauración, apareciendo manifestaciones hemorrágicas y trombóticas. La hemorragia es la manifestación más frecuente en la CID. Puede ser leve o moderada (petequias, equimosis, hematomas, sangrado en lugares de venopunción o herida quirúrgica) o grave con compromiso vital (pulmonar, sistema nervioso central, gastrointestinal o en lecho quirúrgico). La formación de microtrombos de fibrina conduce a la aparición de fallo en diferentes órganos, pudiendo aparecer insuficiencia renal aguda, disfunción hepática por daño hepatocelular por hipoperfusión hepática y trombosis, síndrome de dificultad respiratoria aguda, disfunción del sistema nervioso central, insuficiencia suprarrenal aguda, trombosis venosa profunda, fenómenos tromboembólicos e isquemia miocárdica.
En la CID, debido al consumo de factores de coagulación, se observa un alargamiento de los tiempos de protrombina (TP) y el tiempo parcial de tromboplastina activada (aPTT) en más del 95% de los pacientes con CID. La trombopenia y el descenso progresivo de las mismas evolutivamente es un parámetro sensible de CID.
Otros parámetros utilizados para el diagnóstico de la CID son aquellos que reflejan la activación de la coagulación y la fibrinólisis, y que se ha generado plasmina y trombina, como el DD y los PDF. La elevación de estos marcadores es necesaria para confirmar el diagnóstico.
El principal tratamiento de la CID es el tratamiento de la enfermedad de base que la ha desencadenado. Sin embargo, en ocasiones, es necesario administrar un tratamiento de soporte basado en la restitución de plaquetas y factores de la coagulación y tratamiento anticoagulante. La administración de hemoderivados no debe estar basada en la corrección de alteraciones de laboratorio, sino reservarse a pacientes con sangrado activo o bien a pacientes con alto riesgo de sangrado por procedimientos invasivos.
Las hepatopatías son una causa frecuente de alteración de la coagulación en el paciente pediátrico crítico, apareciendo alteración en las pruebas de coagulación o sangrado manifiesto en el 15% de los pacientes que tienen manifestaciones clínicas o analíticas de disfunción hepática.
La alteración de la hemostasia que aparece en el paciente con hepatopatía es multifactorial, coexistiendo un estado anticoagulante con riesgo de sangrado junto con riesgo de trombosis en estos pacientes.
El hallazgo más frecuente en el enfermo con hepatopatía es el alargamiento del TP. El factor VII es el primero que desciende debido a su corta vida media (4-6h). Según va avanzando la enfermedad, aparece alargamiento del aPTT, reflejando la alteración en la síntesis de otros factores como los dependientes de vitamina K (II, VII, IX y X) y del factor V.
No se recomienda el tratamiento del paciente asintomático solo por las alteraciones en la coagulación, estando el tratamiento reservado a las situaciones con riesgo de sangrado o a las situaciones de sangrado activo. Este tratamiento estará basado en la administración de plasma fresco congelado, plaquetas y vitamina K, usándose recientemente otros agentes, como el factor VII activado recombinante.
Coagulopatías en el paciente pediátrico crítico.
| Coagulopatías con riesgo de sangrado |
| Coagulación intravascular diseminada |
| Enfermedad hepática/insuficiencia hepática |
| Déficit de vitamina K |
| Síndrome de transfusión masiva |
| Sobredosis de anticoagulantes (heparina, anticoagulantes por vía oral) |
| Trombocitopenia (inmunológica, secundaria a fármacos) |
| Defectos plaquetarios adquiridos (fármacos, uremia). |
| Déficits de factores asociados a enfermedades sistémicas: amiloidosis (X), Gaucher (IX), síndrome nefrótico (IX, AT-III) |
| Coagulopatías con riesgo de trombosis |
| Púrpura trombótica trombocitopénica/síndrome hemolítico urémico |
| Trombosis venosa profunda |
| Tromboembolismo pulmonar |
AT-III: antitrombina III.
Diagnóstico diferencial de las coagulopatías más frecuentes en UCIP.
| Situación clínica | Pruebas laboratorio iniciales | Otras pruebas de laboratorio |
| Coagulación intravascular diseminada | Alargamiento de TP, TT, aPTT; descenso de fibrinógeno y plaquetas | Aumento de PDF y DD; disminución de factores V, VII y II (tardío) |
| Transfusión masiva | Alargamiento del TP, aPTT: descenso del fibrinógeno y plaquetas ± alargamiento TT | Descenso de todos los factores; PDF —y DD— (salvo si CID); historia + de transfusión |
| Enfermedad hepática | ||
| Temprana | Alargamiento TP | Descenso del factor VII. |
| Tardía | Alargamiento TP, aPTT, descenso fibrinógeno (terminal) | Descenso de factores II, V, VII, IX, X; descenso de plasminógeno |
| Déficit de vitamina K | Alargamiento del TP, ± alargamiento aPTT (si severo); TT, fibrinógeno y plaquetas normales | Descenso de los factores dependientes de vitamina K; V y VIII normales |
| Sobredosis de anticoagulantes | ||
| Heparina | Alargamiento aPTT y TT | Tiempo de reptilase normal. Corrección con protamina |
| Anticoagulantes orales | Alargamiento del TP, ± alargamiento aPTT (si severo); TT, fibrinógeno y plaquetas normales | Descenso de los factores dependientes de vitamina K; V y VIII normales |
| Púrpura trombótica trombocitopénica | Trombocitopenia, microangiopatía, anemia moderada. TP, aPTT y fibrinógeno normales | ADAMTS 13 déficit/inhibidor, polímeros de vWF; aumento moderado de PDF y DD |
aPTT: tiempo de tromboplastina parcial activado; DD: dímero D; PDF: productos de degradación de la fibrina; TP: tiempo de protrombina; TT: tiempo de trombina; UCIP: Unidad de Cuidados Intensivos Pediátricos; vWF: polímero de factor von Willebrand.
La CID es un síndrome que se caracteriza por una activación incontrolada de la coagulación, lo cual conduce a la formación de trombos de fibrina, que producen oclusión de los vasos de pequeño y mediano calibre, alterando el suministro de sangre a diferentes órganos, pudiendo aparecer fallo multiorgánico2. A su vez, la activación de la coagulación conduce a un consumo de plaquetas y factores de coagulación, pudiendo aparecer hemorragias graves simultáneamente con los fenómenos trombóticos.
EtiologíaLa CID es un trastorno que puede aparecer en diferentes enfermedades (tabla 3). La sepsis es la principal causa de CID en niños3. Tradicionalmente, se ha asociado la CID a la sepsis por gramnegativos, siendo el paradigma la meningococemia (púrpura fulminans) pero se puede asociar a infecciones sistémicas por otras bacterias y por otros microorganismos, como parásitos, virus y hongos. La aparición de CID en la sepsis contribuye a la aparición de fallo multiorgánico y es un predictor de mortalidad. En las infecciones, factores como las endotoxinas o las exotoxinas activan a las células mediadoras de la inflamación que liberan citocinas inflamatorias que activan coagulación. El traumatismo severo, sobre todo el traumatismo craneoencefálico, y otras situaciones en las que se produce daño tisular como grandes quemaduras, crush-injury, hipotermia o grandes cirugías, son otras de las principales causas de CID. La liberación de enzimas tisulares y fosfolípidos por el daño tisular produce la liberación de citocinas proinflamatorias y la activación de la coagulación. La aparición de CID en las primeras horas del traumatismo se asocia a mayor mortalidad. La liberación de enzimas tisulares y fosfolípidos por el daño tisular produce la liberación de citocinas proinflamatorias y la activación de la coagulación. Los tumores sólidos y las neoplasias hematológicas son otra de las causas de CID4. Las leucemias más frecuentes en niños son las agudas linfoblásticas, siendo la CID poco frecuente en este tipo de neoplasias. La CID aparece con más frecuencia en la leucemia promielocítica, en la que se produce una liberación directa de sustancias procoagulantes a la circulación. En los tumores sólidos es más frecuente que aparezca CID crónica, aunque esta manifestación es rara en niños. La aparición de CID en las neoplasias se relaciona principalmente con 2 mecanismos, la expresión de factor tisular por las células tumorales y de una sustancia procoagulante llamada cistina —proteasa que activa el factor X. Otras causas de CID son la reacción transfusional aguda, el síndrome de Kasabah-Merrit y la mordedura de serpiente. Los neonatos, sobre todo los pretérmino, tienen mayor riesgo de desarrollar CID porque tienen un déficit de todos los factores de la coagulación y de factores anticoagulantes como la antitrombina III (AT-III) y sobre todo de la proteína C y S5. Puede aparecer CID en situaciones como complicaciones obstétricas, que producen hipoxia neonatal (abruptio, eclampsia, distrés), la enterocolitis necrosante, el síndrome del distrés respiratorio, la hipotermia y la incompatibilidad rH.
Causas de coagulación intravascular diseminada en niños.
| Infecciones |
| Bacterias: meningococemia, sepsis por grampositivos y gramnegativos |
| Virus: VIH, VVZ, CMV, dengue, virus Ébola |
| Hongos: Candida, Aspergillus |
| Rickettsia |
| Malaria |
| Daño tisular |
| Traumatismo: politraumatismo, traumatismo craneoencefálico grave |
| Crush-injury |
| Gran quemado |
| Cirugías extensas |
| Hipotermia/hipertermia grave |
| Shock e hipoxia grave |
| Pancreatitis aguda |
| Neoplasias |
| Leucemias: leucemia aguda promielocítica, leucemia aguda linfoblástica |
| Linfomas |
| Neuroblastoma |
| Enfermedades hereditarias |
| Déficit de proteína C y S |
| Déficit de antitrombina III |
| Enfermedades gastrointestinales |
| Hepatitis fulminante |
| Síndrome de Reye |
| Causas neonatales |
| Asfixia neonatal |
| Síndrome de distrés respiratorio |
| Enterocolitis necrosante |
| Infecciones neonatales: bacterianas, fúngicas, congénitas (herpes, CMV) |
| Hipotermia |
| Hemólisis masiva (incompatibilidad rH) |
| Miscelánea |
| Hemangioma gigante (síndrome de Kasabach Merrit) |
| Mordedura de serpiente |
| Reacción transfusional aguda |
CMV: citomegalovirus; VIH: virus de la inmunodeficiencia humana; VVZ: virus de la varicela zóster.
La CID se produce por una activación excesiva e incontrolada de la coagulación que conduce a la aparición generalizada de trombos intravasculares. En la CID existe una serie de mecanismos que actúan simultáneamente y que conducen a la activación de la coagulación. La activación del factor tisular, la disminución de la actividad de los anticoagulantes y la inactivación de la fibrinólisis son los principales factores que intervienen en la aparición del estado procoagulante de la CID. El consumo masivo de los factores de la coagulación y de las plaquetas hace que aparezca sangrado simultáneamente a los fenómenos trombóticos2,6 . En el desarrollo de la CID desempeña un papel importante la interrelación entre la activación de la respuesta inflamatoria sistémica y de la coagulación, así citocinas proinflamatorias, como la interleucina-6 (IL-6), intervienen en la iniciación de la coagulación y otras citocinas como el factor de necrosis tumoral alfa (TNF-α) y la IL-1 intervienen en la regulación de los factores anticoagulantes. En los últimos años, se ha visto que esta regulación es bidireccional, y así, diferentes factores de la coagulación como la AT-III, la proteína C y los receptores de las proteasas regulan, a su vez, la respuesta inflamatoria7.
Activación del factor tisularEl fenómeno inicial de la activación de la coagulación mediada por la inflamación es la activación del factor tisular. El factor tisular lo expresan células que no se encuentran en contacto con directo con la sangre. El factor tisular toma contacto con la sangre cuando se rompe la integridad del vaso o cuando las células circulantes lo expresan. En la inflamación, la expresión del factor tisular está mediada por la IL-6. En la sepsis grave, las células mononucleares estimuladas por las citocinas proinflamatorias expresan factor tisular, lo que conduce a una activación de la coagulación7.
Disminución de los factores anticoagulantesLos principales inhibidores de la coagulación (AT-III, proteína C y S) están disminuidos en la CID. Los niveles plasmáticos de AT-III están disminuidos por la combinación de varios factores, como el consumo, la inhibición por medio de la elastasa producida por los neutrófilos activados y por una disminución de la síntesis.
La proteína C activada desempeña un papel importante en la activación de la coagulación en la sepsis por diversos mecanismos8. Existen niveles muy bajos de proteína C por alteración en su síntesis y aumento de su degradación por las elastasas de los neutrófilos activados. Además, la contrarregulación por medio de la trombomodulina está alterada por la activación de la IL-1 y el TNF-α, produciendo una disminución de la actividad de la proteína C.
Inactivación de la fibrinólisisDurante la CID se produce una inactivación de la fibrinólisis. Los principales mediadores de la fibrinólisis durante la inflamación son el TNF-α y la IL-1b. Estos mediadores activan la secreción aumentada y mantenida del factor inhibidor del plasminógeno-1 (PAI-1), por lo q[ue no se produce una degradación de la fibrina y contribuye a la trombosis microvascular.
Microangiopatía trombóticaEn los pacientes críticos, la microangiopatía trombótica puede producir trombosis microvascular y fallo multiorgánico9,10. Aunque la CID y la microangiopatía trombótica son entidades diferenciadas; esta última, cuando es grave, puede producir CID. El ADAMTS 13 (a disentegrin metalloprotease with thrombospodin type 1 modif, number 13) es una proteasa que degrada los multímeros de factor von Willebrand (vWF). Cuando existe una deficiencia de este factor no se degradan los agregados de plaquetas y vWF, produciéndose trombosis intravascular. En diversos estudios se ha observado la disminución de la actividad del ADAMTS13 y la aparición de multímeros de vWF. Estos resultados iniciales sugieren que el ADAMTS 13 podría desempeñar un papel en el desarrollo de la trombosis microvascular y el fallo multiorgánico que aparece en la CID.
Manifestaciones clínicasLas manifestaciones clínicas de la CID van a ser muy variables, dependiendo de la severidad del cuadro y de la velocidad de instauración, apareciendo manifestaciones hemorrágicas y trombóticas. Clásicamente se han descrito 2 formas de presentación de la CID, la forma aguda y la forma crónica. La forma aguda se produce cuando se liberan grandes cantidades de factor tisular al torrente sanguíneo en un periodo corto, produciendo una clínica con fenómenos hemorrágicos y trombóticos con fallo de diversos órganos. La crónica es una forma más larvada, en la que los pacientes van a estar asintomáticos o suelen predominar las complicaciones trombóticas (como trombosis venosa profunda) o sangrado leve. Esta forma de presentación suele aparecer en el paciente oncológico y es muy poco frecuente en niños, por lo que nos referiremos a la aguda.
Clínica hemorrágicaLa hemorragia es la manifestación más frecuente en la CID. Puede ser leve o moderada (petequias, equimosis, hematomas, sangrado en lugares de venopunción o herida quirúrgica) o grave con compromiso vital (pulmonar, sistema nervioso central, gastrointestinal o en lecho quirúrgico). En neonatos, los lugares de sangrado más frecuentes son los sitios de venopunción y la hemorragia gastrointestinal.
Clínica tromboembólicaLa formación de microtrombos de fibrina conduce a la aparición de fallo en diferentes órganos, pudiendo aparecer insuficiencia renal aguda, disfunción hepática por daño hepatocelular por hipoperfusión hepática y trombosis, síndrome de dificultad respiratoria aguda, disfunción del sistema nervioso central, insuficiencia suprarrenal aguda, trombosis venosa profunda, fenómenos tromboembólicos e isquemia miocárdica.
DiagnósticoNo existe una prueba diagnóstica específica para el diagnóstico de la CID. Se basa en la presunción diagnóstica apoyada en diferentes pruebas de laboratorio. En la CID, debido al consumo de factores de coagulación, se observa un alargamiento de los tiempos de protrombina (TP) y el tiempo parcial de tromboplastina activada (aPTT) en más del 95% de los pacientes con CID. El aPTT representa la activación de la vía extrínseca incluido el factor VIII, que es un factor que se encuentra elevado de forma paradójica en la CID, por lo que puede que el aPTT se encuentre en valores normales con prolongación del TP en los pacientes con CID. La trombopenia y el descenso progresivo de las mismas evolutivamente son parámetros sensibles de CID, especialmente en pacientes con sepsis, aunque pueden estar normales en fases tempranas. En esta situación, el descenso progresivo de plaquetas en diferentes medidas es mejor que los valores absolutos. El fibrinógeno disminuye en la CID aunque, como es un reactante de fase aguda, no suele disminuir hasta fases avanzadas de la enfermedad, por lo que es un parámetro con baja sensibilidad (22%)11. En la CID también existe un descenso de los anticoagulantes naturales, como la AT-III y la proteína C. Otros parámetros utilizados para el diagnóstico de la CID son aquellos que reflejan la activación de la coagulación y la fibrinólisis, y la generación de plasmina y trombina, como el dímero D (DD) y los productos de degradación de la fibrina (PDF). La elevación de estos marcadores es necesaria para confirmar el diagnóstico. Además, valores muy elevados de PDF y DD se pueden ver en la CID con hiperfibrinólisis, como la que aparece en la leucemia promielocítica. Sin embargo, en la CID asociada a PAI-1 elevado, se observa una elevación menor de DD y PDF. La elevación del DD y de los PDF tiene una sensibilidad elevada y moderada especificidad para el diagnóstico de CID, ya que estos marcadores se pueden elevar en otras situaciones, como trombosis venosa profunda o traumatismo y cirugía recientes. Además de estas pruebas de laboratorio, existen marcadores moleculares que detectan la generación de trombina (fragmento de protrombina 1 y 2), la generación de trombina y su neutralización (complejo trombina-AT), la activación de la trombina (fibrina soluble, fibrinopéptido A), para la generación de plasmina y su neutralización (complejo plasmina-antiplasmina) y para la activación de plasmina (fibrinopéptido B β15–42). Estos marcadores tienen alta sensibilidad y escasa especificidad. Su utilidad clínica en la actualidad es escasa, ya que son pruebas caras y que no están disponibles en la mayoría de los centros.
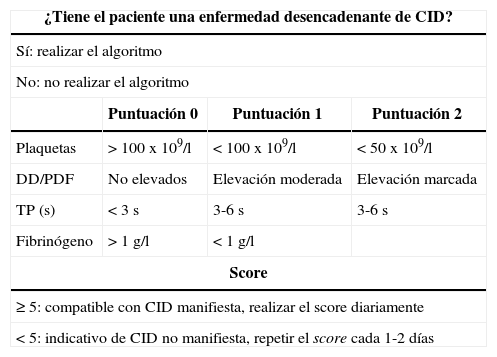
Se han desarrollado diferentes scores clínicos en adultos pare el diagnóstico de CID combinando los hallazgos de laboratorio de manera evolutiva para el diagnóstico de la CID manifiesta y no manifiesta12,13. No se ha estudiado la utilidad de estos scores en niños, si bien existe un estudio que relaciona el score con la mortalidad en pacientes pediátricos sépticos con CID13 (tabla 4).
Score diagnóstico de CID de la International Society of Thrombosis and Hemostasia.
| ¿Tiene el paciente una enfermedad desencadenante de CID? | |||
| Sí: realizar el algoritmo | |||
| No: no realizar el algoritmo | |||
| Puntuación 0 | Puntuación 1 | Puntuación 2 | |
| Plaquetas | > 100x109/l | < 100x109/l | < 50x109/l |
| DD/PDF | No elevados | Elevación moderada | Elevación marcada |
| TP (s) | < 3s | 3-6s | 3-6s |
| Fibrinógeno | > 1g/l | < 1g/l | |
| Score | |||
| ≥ 5: compatible con CID manifiesta, realizar el score diariamente | |||
| < 5: indicativo de CID no manifiesta, repetir el score cada 1-2 días | |||
CID: coagulación intravascular diseminada; DD: dímero D; PDF: productos de degradación de la fibrina; TP: tiempo de protrombina.
El principal tratamiento de la CID es el tratamiento de la enfermedad de base que la ha desencadenado. Sin embargo, en ocasiones, es necesario administrar un tratamiento de soporte basado en la restitución de plaquetas y factores de la coagulación y tratamiento anticoagulante. La administración de hemoderivados no debe estar basada en la corrección de alteraciones de laboratorio, sino reservarse a pacientes con sangrado activo o bien a pacientes con alto riesgo de sangrado por procedimientos invasivos. Se administrarán plaquetas en el paciente con sangrado activo con cifras < 50.000 y en los pacientes que no tienen sangrado con valores entre 10.000 y 20.000, teniendo en cuenta que estas cifras de transfusión se basan en estudios en pacientes con trombopenia secundaria a quimioterapia y no en CID. Se administrará plasma fresco congelado (PFC) al paciente sangrante con alargamiento del TP y aPTT, si bien no hay evidencia de que la administración de plasma frene la activación de la coagulación. Además, se necesitarían grandes cantidades de plasma para conseguir restaurar las cifras de factores de la coagulación. Estará indicado reponer el fibrinógeno bien con crioprecipitado o con concentrado de fibrinógeno cuando exista hipofibrinogenemia con cifras < 50–75mg/dl. La administración de concentrado de factores protrombóticos es discutida en la CID, ya que no aporta todos los factores (no tiene factor V) y puede contener trazas de factores activados que empeoren la CID. Se reservará para aquellos pacientes con importante sobrecarga de volumen. Existen varios estudios sobre la utilización de factor VII recombinante en el niño sangrante, si bien no existen estudios específicos en CID, por lo que no se puede en la actualidad recomendar en CID14.
Basándose en el hecho de que en la CID se produce una activación incontrolada de la coagulación, se ha propuesto diferentes anticoagulantes para el tratamiento de la CID. Existen estudios experimentales en los que se ha observado que la heparina inhibe la activación de la coagulación en la CID, si bien no existen estudios prospectivos aleatorizados al respecto. Su uso estaría indicado en pacientes con CID en los que predomine la trombosis con tromboembolismo arterial o venoso, púrpura fulminans severa con isquemia acra11. Como existe, a su vez, alto riesgo de sangrado, se utilizaría heparina sódica en perfusión continua por su vida media corta y su potencial reversibilidad. Estudios con otros anticoagulantes, como el factor inhibidor del factor tisular, en pacientes con CID mostraron resultados prometedores en pacientes con sepsis, si bien no se han comprobado en el ensayo en fase III15. La AT III se ha utilizado en pacientes adultos con CID. Su administración se ha relacionado con una mejoría de los parámetros de laboratorio, si bien no se ha demostrado un descenso de la mortalidad16. La escasa evidencia sobre su uso hace que este no esté recomendado. Se ha postulado que el uso de proteína C activada podría restaurar la anticoagulación y reducir la severidad de la CID. Se han realizado diferentes estudios en niños y en adultos con proteína C recombinante (drotrecogin α) en pacientes con sepsis severa (con o sin CID)17. En alguno de ellos se observaba disminución de la mortalidad con una mayor incidencia de sangrado. En la población pediátrica no se han demostrado estos hallazgos y el riesgo de sangrado es mayor, por lo que no estaría indicado su uso en el paciente pediátrico18–20. No está indicado el uso de fármacos antifibrinolíticos en la CID, salvo en los pacientes con CID con hiperfibrinólisis asociada a leucemia promielocítica, en los que se podría usar el ácido tranexámico. En los niños trombocitopenia asociada a fallo multiorgánico y descenso del ADASMTS13 estaría indicada la realización de recambio plasmático21.
Coagulopatía asociada a la enfermedad hepáticaLas hepatopatías son una causa frecuente de alteración de la coagulación en el paciente pediátrico crítico, apareciendo alteración en las pruebas de coagulación o sangrado manifiesto en el 15% de los pacientes que tienen manifestaciones clínicas o analíticas de disfunción hepática22,23.
PatogeniaLa alteración de la hemostasia que aparece en el paciente con hepatopatía es multifactorial, coexistiendo un estado anticoagulante con riesgo de sangrado junto con riesgo de trombosis en estos pacientes24,25. En la enfermedad hepática se produce una alteración de la síntesis de múltiples proteínas plasmáticas, incluyendo los factores de la coagulación II, V, VII, IX y X. La síntesis de fibrinógeno se mantiene hasta estadios más avanzados de la enfermedad. Las plaquetas suelen estar disminuidas en los pacientes con hepatopatía, sobre todo en los pacientes con hipertensión portal, si bien los niveles altos de vWF y el descenso del ADAMTS-13 en estos podrían contrarrestar este efecto. También aparece un aumento de la fibrinólisis, observándose un aumento de los niveles de activador del plasminógeno y del thrombin activated fibrinolysis inhibitor. Los pacientes con enfermedad hepática también pueden presentar un descenso de las proteínas anticoagulantes dependientes de vitamina K, como la proteína C, la proteína S y la AT-III. Este hecho, junto con los niveles elevados de factor VIII y vWF, hacen que estos pacientes presenten también mayor riesgo de trombosis26. Como coexisten tanto alteraciones que predisponen al sangrado como alteraciones que predisponen a la trombosis, se cree que en la tendencia al sangrado que aparece en el paciente con hepatopatía terminal desempeñan un papel importante otras condiciones, como las alteraciones hemorrágicas secundarias a la hipertensión portal, la disfunción endotelial, las infecciones bacterianas y la insuficiencia renal27 (tabla 5).
Patrones de alteraciones procoagulantes y anticoagulantes en los pacientes con hepatopatía crónica en cada fase de la hemostasia.
| Fase de la hemostasia | Alteraciones procoagulantes | Alteraciones anticoagulantes |
| Hemostasia primaria | Aumento vWF, disminución ADAMTS-13 | Disminución de plaquetas |
| Coagulación | Descenso anticoagulantes antitrombina III y proteínas C y S | Descenso de fibrinógeno y factores II, V, VII, IX, X, XI |
| Fibrinólisis | Plasminógeno disminuido, aumento de PAI | Aumento t-PA, descenso TAFI, descenso del inhibidor del plasminógeno |
PAI: factor inhibidor del plasminógeno; TAFI: thrombin activated fibrinolysis inhibitor; t-PA: activador del plasminógeno; vWF: factor von Willebrand.
El hallazgo más frecuente en el enfermo con hepatopatía es el alargamiento del tiempo de protrombina. El factor VII es el primero que desciende debido a su corta vida media (4–6h). Según va avanzando la enfermedad aparece alargamiento del aPTT, reflejando la alteración en la síntesis de otros factores como los dependientes de vitamina K (II, VII, IX y X) y del factor V. Los niveles de fibrinógeno se suelen mantener estables hasta fase avanzadas de la enfermedad. El aumento de la fibrinólisis hace que aumenten el DD y los PDF, siendo difícil el diagnóstico diferencial entre la fibrinólisis de la hepatopatía y la existencia de CID concomitante. El recuento plaquetario suele estar disminuido. Está en estudio la utilidad de otros métodos de diagnóstico en estos pacientes, como el tromboelastograma y los test de generación de trombina, para poder evaluar el riesgo procoagulante en estos pacientes, si bien su uso aún no está generalizado.
TratamientoNo se recomienda el tratamiento del paciente asintomático solo por las alteraciones en la coagulación, estando el tratamiento reservado a las situaciones con riesgo de sangrado o a las situaciones de sangrado activo. El PFC se puede usar de forma profiláctica o como tratamiento cuando existe sangrado activo. Se administrará PFC en el paciente no sangrante con alteración de los tiempos de coagulación cuando vayan a ser sometidos a procedimientos invasivos, teniendo como umbral PT/aPTT > 1,5, si bien existe poca evidencia sobre esta indicación. El uso de plasma fresco de forma profiláctica no ha demostrado un descenso del riesgo de sangrado o mejoría del pronóstico. A su vez, existe poca evidencia sobre la dosificación del plasma, tanto en adultos como en niños; las últimas guías británicas recomiendan dosis 15ml/kg (10–20ml/kg)22. La administración de plasma no está exenta de riesgo, pudiendo aparecer lesión pulmonar aguda (TRALI), sobrecarga de volumen, reacciones alérgicas y transmisión de infecciones. Se administrará vitamina K a los pacientes con enfermedad hepática, asumiendo que en estos pacientes los niveles de vitamina K están disminuidos, si bien la respuesta a la vitamina K en la hepatopatía grave es muy pobre. La dosis es 0,3mg/kg, máxima 10mg por vía intravenosa u oral. Se administrará fibrinógeno con valores por debajo de 100mg/dl. Se puede administrar en forma de crioprecipitado o en forma de concentrado de fibrinógeno. La dosis de crioprecipitado es de 4–5ml/kg. El concentrado de fibrinógeno se utilizó originariamente en los pacientes con hipofibrinogenemia congénita. Su uso en hipofibrinogenemia adquirida está ampliamente extendido, aunque en la actualidad no existe suficiente evidencia científica para poder generalizar su uso.
La transfusión de plaquetas se realizará si están por debajo de 50.000 en el paciente con sangrado activo o a aquel que va a ser sometido a un procedimiento invasivo. El factor VII activado se usa en situaciones de sangrado grave refractario al tratamiento con plasma y plaquetas. El uso de factor VII activado ha aumentado significativamente en niños y en adultos en los últimos años28. Existen escasos estudios que demuestran su eficacia en el control del sangrado, en los que no se observa una reducción de la mortalidad. La mayor parte de los estudios comparan el factor VIIa con placebo, en lugar de con plasma o concentrado de factores protrombóticos29. Estudios en neonatos indican también una mayor incidencia de trombosis; sin embargo, esta no es mayor a la que se produciría con el tratamiento con PFC28. Se podría concluir que el factor VIIa es eficaz en el control del sangrado, pudiendo estar indicado en los pacientes con sangrado incontrolable a pesar de la transfusión de plasma y plaquetas, y en aquellos pacientes con sangrado y sobrecarga de volumen tras una dosis inicial de plasma30. El complejo de factores protrombóticos contiene los factores II, VII, IX y X junto con proteína C y S, y trazas de AT. La mayor parte de las limitaciones que existen con respecto al uso de factor VIIa existen con respecto al uso del complejo de factores protrombóticos, por la falta de estudios prospectivos aleatorizados al respecto. Actualmente, podría estar indicado en el paciente con hemorragia grave que no se controla con las medidas habituales y en aquellos con sobrecarga de volumen y como preparación para la cirugía electiva o el trasplante en el paciente con enfermedad hepática grave31. Por último, no se ha comprobado la utilidad de los fármacos antifibrinolíticos en el paciente pediátrico con coagulopatía asociada a hepatopatía.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.