













Puntos clave

Las anomalías de la diferenciación sexual (ADS) constituyen un amplio abanico de procesos patológicos originados por alguna anomalía en alguna de las etapas del desarrollo fetal imprescindibles para el desarrollo normal del sexo genético (cariotipo, gonosomas), del sexo gonadal (ovarios o testículos) y/o del sexo genital interno y/o externo (masculino o femenino). Su frecuencia es baja e inferior a 1/2.000 recién nacidos, aunque variable según las etiologías, por lo que se incluyen actualmente dentro de la definición de las “enfermedades raras”, entendidas como poco frecuentes. Su etiología es genética y monogénica en su mayor proporción, habiéndose clonado y descrito unos 32-40 genes en la cascada de proteínas necesarias para una normal diferenciación femenina o masculina. A pesar de los avances alcanzados a lo largo de los últimos 20 años, algunos casos quedan aún sin diagnóstico etiológico definido, sea por falta de estudio molecular o a la espera de la descripción de un nuevo gen.
Fundamentos genéticos y hormonales de la diferenciación sexualLa determinación del sexo genético tiene lugar en el momento de la fecundación, mientras que la diferenciación de los sexos gonadal y genital se produce durante períodos críticos de la vida fetal1. Sus etapas, su regulación génica y las proteínas y esteroides implicados en la diferenciación sexual han sido objeto de numerosos estudios a lo largo de la segunda mitad del siglo xx, sin embargo prosiguen las investigaciones a principios del siglo xxi. Cada etapa está sujeta a posibles alteraciones que pueden condicionar anomalías en todos o en alguno de los tres niveles de diferenciación sexual: el cromosómico, el gonadal o el genital, dando lugar a los denominados “trastornos o anomalías de la diferenciación sexual”. Cuando éstos comportan una diferenciación genital externa ambigua o discordante con el sexo genético o gonadal, pueden ser denominadas “estados intersexuales”2.
Lectura rápida

La diferenciación sexual durante el período fetal se produce a 3 niveles, hasta ahora bien descritos: el genético (cariotipo: femenino 46,XX o masculino 46,XY), el gonadal (gónada femenina: ovario o gónada masculina: testículo) y el genital (genitales internos y externos: femeninos o masculinos). Depende de una cascada de genes cuyos productos (proteínas y hormonas esteroideas) intervienen en la diferenciación de las gónadas y los genitales.

Las anomalías de la diferenciación sexual (ADS), comporten o no un “estado intersexual”, se han clasificado de diversas formas. En este artículo se sigue la clasificación derivada del consenso internacional alcanzado en el año 20063 sobre nomenclatura y clasificación que se presenta en la tabla 1. Esta primera clasificación está basada en el cariotipo, de modo que se distinguen 3 grandes tipos de ADS: cuando existen anomalías en los cromosomas sexuales (en la tabla 1 se mencionan los más frecuentes y se evidencia que no en todos los casos existe un estado intersexual, así es en los síndromes de Turner y de Klinefelter), cuando el cariotipo es femenino 46,XX y, finalmente, cuando el cariotipo es masculino 46,XY. Es importante recalcar que, en este consenso3 han desaparecido varios términos anteriormente utilizados: “hermafroditismo verdadero” ha sido sustituido por “quimera o ADS ovotesticular”, el “seudohermafroditismo femenino” por “ADS 46,XX” y el “seudohermafroditismo masculino” por “ADS 46,XY”.
Clasificación de las anomalías de la diferenciación sexual
ADS con anomalías de los cromosomas sexuales
|
| ADS con cariotipo 46,XX (anteriormente seudohermafrodistismo femenino) |
| ADS con cariotipo 46,XY (anteriormente seudohermafrodistimo masculino) |
ADS: anomalías de la diferenciación sexual.
A su vez, las ADS con cariotipo 46,XX se subclasifican (tabla 2) según las etiologías en anomalías del desarrollo ovárico, en virilización por la presencia de un exceso de andrógenos durante la vida fetal con las diferentes etiologías y finalmente algunas malformaciones múltiples. Las ADS con cariotipo 46,XY se clasifican también según las etiologías (tabla 3) en anomalías primarias del desarrollo testicular con los diferentes genes implicados hasta ahora conocidos, en anomalías de la síntesis o de la acción de los andrógenos con los diferentes genes implicados, en anomalías de la síntesis o de la acción del factor inhibidor de los conductos de Müller y en otros síndromes que asocian malformaciones múltiples.
Etiología de las anomalías de la diferenciación sexual con cariotipo 46,XX
Anomalías del desarrollo gonadal (ovario)
|
Exceso de andrógenos
|
Otros
|
Etiología de las anomalías de la diferenciación sexual con cariotipo 46,XY
Anomalías del desarrollo gonadal (testículo)
|
Regresión testicular
|
Otros
|
El diagnóstico de las ADS es pluridisciplinario y requiere primero la conjunción de los estudios clínicos (antecedentes personales y familiares, exploración clínica y de imagen) y bioquímicos (bioquímica general y análisis hormonales) a los que debe añadirse la determinación del cariotipo. A su vez, y en función del cariotipo y del análisis de los datos clínicos y bioquímicos, se podrá analizar uno o varios genes candidatos, a condición de que se haya conseguido una orientación clara del diagnóstico etiológico (fig. 1).


La primera etapa con posibles anomalías es la del sexo genético o cromosómico. Su consecuencia serán anomalías en la segunda etapa de la diferenciación sexual, la gonadal, acompañadas o no de anomalías en la tercera etapa de la diferenciación sexual, la genital interna y/o externa. Dentro de este capítulo figuran las disgenesias gonadales tipo síndrome de Turner con cariotipo 45,X y otros, las disgenesias gonadales mixtas con mosaicos 45,X/46,XY y el quimerismo ovotesticular con mosaico 46,XX/46,XY. La determinación del cariotipo permite pues la clasificación del paciente en uno de los 3 grandes apartados de la clasificación actual (tabla 1).
Cuando el cariotipo es 46,XX o 46,XY pero la diferenciación gonadal y/o la genital son discordantes con el sexo genético, las terminologías que se aplican son de ADS con cariotipo 46,XX (tabla 2) o de ADS con cariotipo 46,XY (tabla 3).
Lectura rápida
Las anomalías de la diferenciación sexual (ADS) constituyen un amplio abanico de procesos patológicos originados por alguna anomalía en alguna de las etapas del desarrollo fetal imprescindibles para el desarrollo normal del sexo genético (cariotipo, gonosomas), del sexo gonadal (ovarios o testículos) y/o del sexo genital interno y/o externo (masculino o femenino). Su frecuencia es baja e inferior a 1/2.000 recién nacidos, aunque variable según las etiologías, por lo que se incluyen actualmente dentro de la definición de las “enfermedades raras”, entendidas como “poco frecuentes”. Su etiología es genética y monogénica en su mayor proporción, habiéndose clonado y descrito unos 32-40 genes en la cascada de proteínas necesarias para una normal diferenciación femenina o masculina.
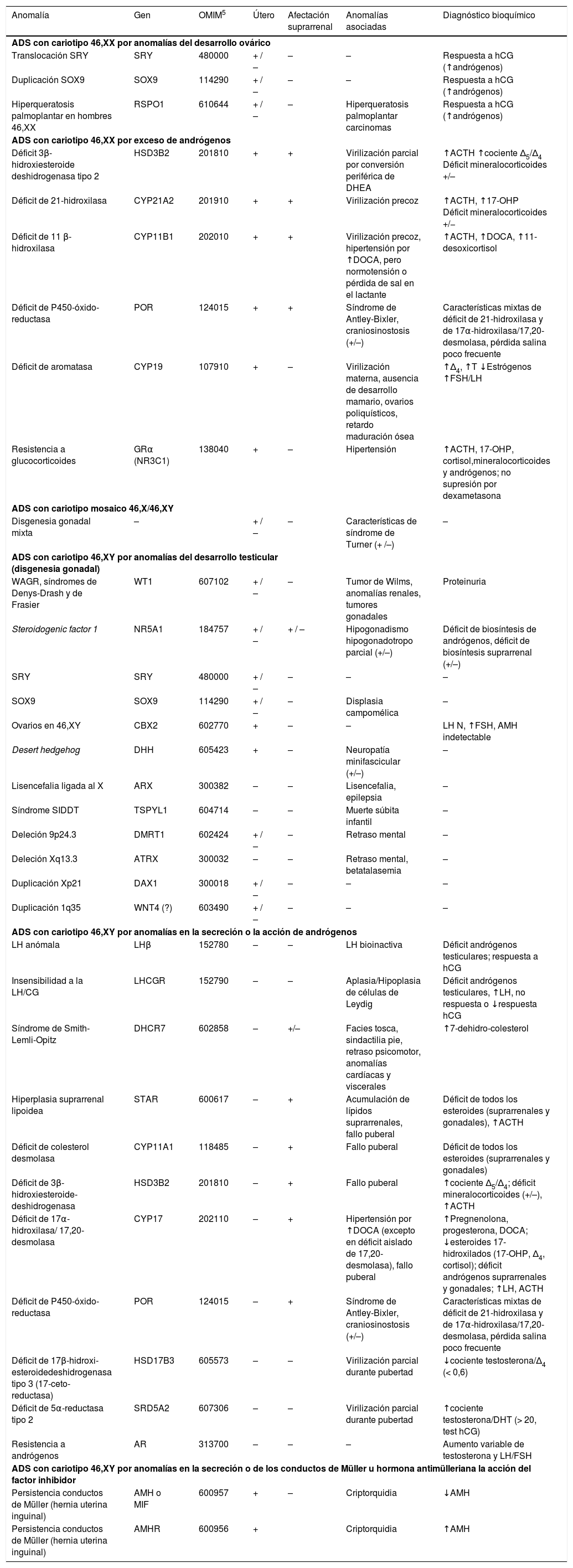
Actualmente está bien establecido que la etiología de un número importante de ADS son mutaciones en genes necesarios para la diferenciación sexual normal. En la tabla 4 se resumen las ADS de causa monogénica o cromosómica conocida con sus características fenotípicas y bioquímicas. La demostración de la mutación o mutaciones establece el diagnóstico etiológico definitivo y permite los estudios familiares, el consejo genético y el diagnóstico prenatal. Para ello es necesario utilizar técnicas de biología molecular, entre las cuales la secuenciación automática de fragmentos amplificados por PCR facilita enormemente la obtención de resultados rápidos. Las mutaciones detectadas pueden ir desde la deleción total del gen, que a veces incluye material genético contiguo, pasando por deleciones parciales (desde varios exones hasta una sola base), hasta cambios puntuales de un solo nucleótido que puede provocar desde una parada en la pauta de lectura hasta una proteína con estructura anómala y pérdida de función. Los resultados obtenidos en los defectos estructurales y en las secuencias se comparan con las secuencias registradas en las bases de datos génicos del National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov).
Anomalías de la diferenciación sexual de causa monogénica o cromosómica conocida con sus características fenotípicas y bioquímicas
| Anomalía | Gen | OMIM5 | Útero | Afectación suprarrenal | Anomalías asociadas | Diagnóstico bioquímico |
|---|---|---|---|---|---|---|
| ADS con cariotipo 46,XX por anomalías del desarrollo ovárico | ||||||
| Translocación SRY | SRY | 480000 | + / – | – | – | Respuesta a hCG (↑andrógenos) |
| Duplicación SOX9 | SOX9 | 114290 | + / – | – | – | Respuesta a hCG (↑andrógenos) |
| Hiperqueratosis palmoplantar en hombres 46,XX | RSPO1 | 610644 | + / – | – | Hiperqueratosis palmoplantar carcinomas | Respuesta a hCG (↑andrógenos) |
| ADS con cariotipo 46,XX por exceso de andrógenos | ||||||
| Déficit 3β-hidroxiesteroide deshidrogenasa tipo 2 | HSD3B2 | 201810 | + | + | Virilización parcial por conversión periférica de DHEA | ↑ACTH ↑cociente Δ5/Δ4 Déficit mineralocorticoides +/– |
| Déficit de 21-hidroxilasa | CYP21A2 | 201910 | + | + | Virilización precoz | ↑ACTH, ↑17-OHP Déficit mineralocorticoides +/− |
| Déficit de 11 β-hidroxilasa | CYP11B1 | 202010 | + | + | Virilización precoz, hipertensión por ↑DOCA, pero normotensión o pérdida de sal en el lactante | ↑ACTH, ↑DOCA, ↑11-desoxicortisol |
| Déficit de P450-óxido-reductasa | POR | 124015 | + | + | Síndrome de Antley-Bixler, craniosinostosis (+/–) | Características mixtas de déficit de 21-hidroxilasa y de 17α-hidroxilasa/17,20-desmolasa, pérdida salina poco frecuente |
| Déficit de aromatasa | CYP19 | 107910 | + | – | Virilización materna, ausencia de desarrollo mamario, ovarios poliquísticos, retardo maduración ósea | ↑Δ4, ↑T ↓Estrógenos ↑FSH/LH |
| Resistencia a glucocorticoides | GRα (NR3C1) | 138040 | + | – | Hipertensión | ↑ACTH, 17-OHP, cortisol,mineralocorticoides y andrógenos; no supresión por dexametasona |
| ADS con cariotipo mosaico 46,X/46,XY | ||||||
| Disgenesia gonadal mixta | – | + / – | – | Características de síndrome de Turner (+ /–) | – | |
| ADS con cariotipo 46,XY por anomalías del desarrollo testicular (disgenesia gonadal) | ||||||
| WAGR, síndromes de Denys-Drash y de Frasier | WT1 | 607102 | + / – | – | Tumor de Wilms, anomalías renales, tumores gonadales | Proteinuria |
| Steroidogenic factor 1 | NR5A1 | 184757 | + / – | + / – | Hipogonadismo hipogonadotropo parcial (+/–) | Déficit de biosíntesis de andrógenos, déficit de biosíntesis suprarrenal (+/–) |
| SRY | SRY | 480000 | + / – | – | – | – |
| SOX9 | SOX9 | 114290 | + / – | – | Displasia campomélica | – |
| Ovarios en 46,XY | CBX2 | 602770 | + | – | – | LH N, ↑FSH, AMH indetectable |
| Desert hedgehog | DHH | 605423 | + | – | Neuropatía minifascicular (+/–) | – |
| Lisencefalia ligada al X | ARX | 300382 | – | – | Lisencefalia, epilepsia | – |
| Síndrome SIDDT | TSPYL1 | 604714 | – | – | Muerte súbita infantil | – |
| Deleción 9p24.3 | DMRT1 | 602424 | + / – | – | Retraso mental | – |
| Deleción Xq13.3 | ATRX | 300032 | – | – | Retraso mental, betatalasemia | – |
| Duplicación Xp21 | DAX1 | 300018 | + / – | – | – | – |
| Duplicación 1q35 | WNT4 (?) | 603490 | + / – | – | – | – |
| ADS con cariotipo 46,XY por anomalías en la secreción o la acción de andrógenos | ||||||
| LH anómala | LHβ | 152780 | – | – | LH bioinactiva | Déficit andrógenos testiculares; respuesta a hCG |
| Insensibilidad a la LH/CG | LHCGR | 152790 | – | – | Aplasia/Hipoplasia de células de Leydig | Déficit andrógenos testiculares, ↑LH, no respuesta o ↓respuesta hCG |
| Síndrome de Smith-Lemli-Opitz | DHCR7 | 602858 | – | +/– | Facies tosca, sindactilia pie, retraso psicomotor, anomalías cardíacas y viscerales | ↑7-dehidro-colesterol |
| Hiperplasia suprarrenal lipoidea | STAR | 600617 | – | + | Acumulación de lípidos suprarrenales, fallo puberal | Déficit de todos los esteroides (suprarrenales y gonadales), ↑ACTH |
| Déficit de colesterol desmolasa | CYP11A1 | 118485 | – | + | Fallo puberal | Déficit de todos los esteroides (suprarrenales y gonadales) |
| Déficit de 3β-hidroxiesteroide-deshidrogenasa | HSD3B2 | 201810 | – | + | Fallo puberal | ↑cociente Δ5/Δ4; déficit mineralocorticoides (+/–), ↑ACTH |
| Déficit de 17α-hidroxilasa/ 17,20-desmolasa | CYP17 | 202110 | – | + | Hipertensión por ↑DOCA (excepto en déficit aislado de 17,20-desmolasa), fallo puberal | ↑Pregnenolona, progesterona, DOCA; ↓esteroides 17-hidroxilados (17-OHP, Δ4, cortisol); déficit andrógenos suprarrenales y gonadales; ↑LH, ACTH |
| Déficit de P450-óxido-reductasa | POR | 124015 | – | + | Síndrome de Antley-Bixler, craniosinostosis (+/–) | Características mixtas de déficit de 21-hidroxilasa y de 17α-hidroxilasa/17,20-desmolasa, pérdida salina poco frecuente |
| Déficit de 17β-hidroxi-esteroidedeshidrogenasa tipo 3 (17-ceto-reductasa) | HSD17B3 | 605573 | – | – | Virilización parcial durante pubertad | ↓cociente testosterona/Δ4 (< 0,6) |
| Déficit de 5α-reductasa tipo 2 | SRD5A2 | 607306 | – | – | Virilización parcial durante pubertad | ↑cociente testosterona/DHT (> 20, test hCG) |
| Resistencia a andrógenos | AR | 313700 | – | – | – | Aumento variable de testosterona y LH/FSH |
| ADS con cariotipo 46,XY por anomalías en la secreción o de los conductos de Müller u hormona antimülleriana la acción del factor inhibidor | ||||||
| Persistencia conductos de Müller (hernia uterina inguinal) | AMH o MIF | 600957 | + | – | Criptorquidia | ↓AMH |
| Persistencia conductos de Müller (hernia uterina inguinal) | AMHR | 600956 | + | Criptorquidia | ↑AMH | |
Es muy importante subrayar que el análisis de genes candidatos a ser la causa de una ADS sólo está justificado cuando los diagnósticos clínico y bioquímico han podido orientar adecuadamente el estudio (fig. 1).
Es muy probable que en un futuro próximo se disponga de microarrays diseñados para el diagnóstico de anomalías en varios de los genes que regulan la diferenciación sexual. Ello facilitará enormemente el trabajo de los laboratorios de diagnóstico molecular.
Cuadros clínicos más relevantesAnomalías de la diferenciación sexual con anomalías de los cromosomas sexuales- —
Cariotipo 45,X y mosaico 45,X/46,XX: síndrome de Turner y variantes. La mayor parte de las anomalías de la diferenciación ovárica no modifican la diferenciación genital femenina y provocan, en el sexo femenino, un hipogonadismo primario.
- —
Cariotipo 47,XXY: síndrome de Klinefelter y variantes. En el síndrome de Klinefelter rara vez existe ambigüedad de los genitales externos, pero sí es frecuente la ginecomastia a partir de la pubertad. Los síndromes con múltiples X o Y rara vez asocian hipospadias, pene hipoplásico y criptorquidia.
- —
Mosaico 45,X/46,XY: disgenesias gonadales mixtas. El diagnóstico de disgenesia gonadal mixta (DGM) o disgenesia gonadal asimétrica se aplica a un grupo heterogéneo de pacientes que presentan ambigüedad de los genitales externos y cuyas gónadas, asimétricas, consisten en un testículo con grados variables de disgenesia en un lado y una gónada fibrosa, indiferenciada, en el otro. Muchos presentan anomalías gonosómicas, predominando el cariotipo 45,X/46,XY, pero el cariotipo puede ser 45,X o 46,XY. Algunos pacientes, y más los mosaicos 45,X/46,XY, presentan características fenotípicas del síndrome de Turner, incluidos la talla baja y, con mucha menor frecuencia, malformaciones renales y cardíacas.
- —
Mosaico 46,XX/46,XY: quimera ovotesticular. La quimera ovotesticular (anteriormente denominada hermafroditismo verdadero) consiste teóricamente en la presencia, en un mismo individuo, de los 2 tipos de gónada, testículo y ovario, cuyas estructuras deben estar bien diferenciadas y cuya función debiera ser normal. En la práctica, el diagnóstico es anatomopatológico y el fenotipo y el genotipo son variables. El genotipo más frecuente es 46,XX, pero también puede contener un cromosoma Y, pudiendo entonces ser 46,XY o el mosaico 46,XX/46,XY.
La clasificación actual de las ADS que se acompañan de cariotipo 46,XX se presenta en la tabla 2. El primer apartado incluye todas las posibles anomalías de diferenciación de la gónada femenina, el ovario, mientras que en el segundo se presentan todas las causas de virilización de los genitales femeninos cuando la gónada presenta una diferenciación normal en ovario y el tercer apartado incluye malformaciones urogenitales múltiples que simulan una virilización. En la tabla 4 se resumen las ADS de causa monogénica o cromosómica conocida con sus características fenotípicas y bioquímicas, entre ellas las que llevan el cariotipo 46,XX. Las características clínicas, bioquímicas y moleculares y el tratamiento de la forma más frecuente (la hiperplasia suprarrenal congénita, HSC) pueden ser revisados en Audí et al2 y Sánchez Bachega y Bilharino de Mendonça4.
Anomalías del desarrollo gonadal (ovario)Disgenesia gonadal46,XXLas pacientes presentan fenotipo femenino, cintillas gonadales, infantilismo sexual, talla normal o alta, ausencia de anomalías congénitas presentes en el síndrome de Turner y cariotipo 46,XX. Algunos casos podrían deberse a deleciones de la zona distal del brazo corto del cromosoma 9 en cuyo locus 9p24.3 están situados los genes DMRT1 y DMRT2 (OMIM 602424 y OMIM 604935)5. Otro gen candidato sería el del factor de crecimiento BMP15 (bone morphogenic protein 15), también denominado GDF9B (growth differentiation factor 9B) (OMIM 300247)5,6. También las mutaciones inactivadoras del gen del receptor de la FSH (FSHR) en 2p21-p16 provocan disgenesia ovárica con hipogonadismo hipergonadotropo (OMIM 233300)5, así como las que afectan al gen FOXL2 en 3q23 en el marco del síndrome BPES (blefarofimosis, ptosis y epicanto inverso con insuficiencia ovárica e infertilidad (OMIM 110100)5.
Quimera ovotesticular 46,XXEn el apartado de ADS con anomalías cromosómicas se ha mencionado el quimerismo ovotesticular que comporta la diferenciación de las gónadas en ovario y testículo (anteriormente denominado hermafroditismo verdadero). En estos casos el cariotipo más frecuente es el 46,XX, desconociéndose los mecanismos moleculares implicados en el desarrollo anómalo.
Desarrollo testicular con cariotipo 46,XXLa detección de un cariotipo 46,XX en un varón con virilización normal de los genitales externos puede ser un hallazgo casual, pero suele darse en el estudio de alguna anomalía, como criptorquidia unilateral o bilateral, ginecomastia al llegar a la pubertad, infertilidad o, más rara vez, casos familiares que pueden incluir infertilidad, disgenesia gonadal mixta (DGM) y una quimera ovotesticular. Algunos pueden presentar ambigüedad de los genitales externos (hasta un 15-20%).
Los mecanismos por los cuales, a pesar del cariotipo 46,XX, las gónadas se han diferenciado en testes parecen poder ser varios. En algunos casos se detectan porciones del brazo corto del cromosoma Y translocadas sobre el cromosoma X o un autosoma siempre conteniendo el gen SRY, mientras que en otros esa diferenciación parece depender de genes autosómicos dominantes o ligados al cromosoma X.
Recientemente se ha comenzado a describir un efecto de dosis génica para algunos de los otros genes que intervienen en la cascada génica reguladora de la diferenciación del testículo: así, la duplicación de un fragmento del cromosoma 17 que contiene el gen SOX-9 (OMIM 114290)5 podría haber provocado una diferenciación parcial de las gónadas a testículo en un paciente con cariotipo 46,XX.
En el apartado dedicado a las disgenesias gonadales 46,XY se menciona el efecto de dosis del gen WNT4 según el cual en el ratón XX con los 2 alelos de WNT4 anulados (WNT4–/–) en la gónada primitiva se diferencian células de Leydig, aunque el testículo adulto es disgenético7. Este efecto no ha sido demostrado en los seres humanos, pero sí el efecto de aumento de dosis (duplicación de un alelo, con ADS en los 46,XY)8. Las mutaciones inactivadoras del gen RSPO1 (OMIM 610644)5 facilitarían el desarrollo testicular en los 46,XX.
Anomalías de la diferenciación genital con cariotipo 46,XX por exceso de andrógenosEl diagnóstico comporta que los sexos genético y gonadal son femeninos normales (cariotipo 46,XX y presencia de ovarios bien diferenciados), pero el sexo genital es ambiguo o totalmente virilizado. El origen de los andrógenos que virilizan al feto femenino puede ser fetal, fetoplacentario o exclusivamente materno (tabla 2).
De origen fetal- —
Hiperplasia suprarrenal congénita. La causa más frecuente es de origen fetal y entre ellas la principal es la hiperplasia suprarrenal congénita (HSC), siendo también la causa más frecuente de ambigüedad genital o estado intersexual o ADS. Existen 4 déficit enzimáticos suprarrenales que provocan una virilización del feto femenino: los déficit de 21-hidroxilasa, de 11β-hidroxilasa, de 3β-hidroxiesteroidedeshidrogenasa y de P450 óxido-reductasa. El más frecuente es el déficit de 21-hidroxilasa. En cada caso el gen candidato viene orientado por la clínica y las determinaciones hormonales y el análisis molecular deberá demostrar la presencia de mutaciones bialélicas ya que las mutaciones en los genes CYP21A2, CYP11B1, HSD3B2 o POR son autosómicas con efecto recesivo4,9–11.
- —
Tumores fetales productores de andrógenos. Existen muy pocos casos publicados, todos ellos debidos a un tumor suprarrenal congénito, no habiéndose descrito ningún tumor ovárico congénito virilizante de un feto femenino. Puede tratarse de un carcinoma o de un adenoma benigno.
- —
Mutaciones del receptor de glucocorticoides. Las mutaciones inactivadoras del receptor de glucocorticoides (gen GR o NR3C1 en OMIM 138040)5 provocan un gran aumento de secreción de ACTH así como del cortisol y de sus precursores y entre ellos de precursores de andrógenos, como androstenodiona y otros, que se transforman a nivel periférico en testosterona. Se han descrito casos de virilización tanto posnatal como prenatal.
- —
Déficit de aromatasa. La enzima aromatasa (P-450aro) es el producto del gen CYP19, localizado en el cromosoma 15 (OMIM 107910)5. El estudio clínico y hormonal de varios casos de déficit de aromatasa (P-450aro) por mutaciones en homocigosis o doble heterocigosis del gen CYP19 ha demostrado que la aromatasa sólo es expresada en la placenta por células de origen fetal, manifestándose el déficit en fetos homocigotos o heterocigotos dobles. Se pone de manifiesto que el gran aumento de producción de estrógenos durante el embarazo probablemente no sea necesario para ninguna función importante, como no sea para disminuir los andrógenos suprarrenales fetales que virilizarían al feto femenino.
- —
Déficit de óxido-reductasa. El déficit de P-450 óxido-reductasa (gen POR, OMIM 124015)5 afecta a todas las actividades enzimáticas microsomales que necesitan esta flavoproteína donadora de electrones. Entre las enzimas de la esteroidogénesis afecta a las actividades de CYP17 y de CYP21, por lo tanto provoca un déficit combinado de 21-hidroxilasa y de 17α-hidroxilasa/17,20-desmolasa. En el sexo genético y gonadal femenino provoca una clínica parecida a la del déficit de 21-hidroxilasa a la que se suman características fenotípicas similares a las del síndrome de Antley-Bixler (OMIM 207410)5. Se han descrito diversas mutaciones en homocigosis o heterocigosis compuesta9,10.
- —
Tumores maternos virilizantes. Es una causa también poco frecuente de virilización del feto femenino, aunque lo es más que el caso anterior de tumor fetal virilizante. El hiperandrogenismo ovárico materno puede ser debido a: tumores ováricos secretores de andrógenos (tumores de las células de la granulosa o tumores de células de Leydig-Sertoli), tumores ováricos con activación de la estroma (tumor de Krukenberg y otros), luteoma del embarazo por luteinización ovárica como respuesta a la hCG, siendo este último el tumor ovárico más frecuentemente productor de virilización fetal de origen materno.
- —
Hiperplasia suprarrenal materna incorrectamente tratada. Rara vez provoca en el feto femenino una virilización, excepto si éste sufre la misma enfermedad. Además, la HSC, sobre todo en su forma clásica, suele ser causa de infertilidad si no es tratada de forma adecuada.
- —
Fármacos androgénicos.
- —
Malformaciones múltiples urogenitales sin etiología hormonal. Las ADS de los genitales internos femeninos derivados de los conductos de Müller (trompas, útero y dos tercios superiores de la vagina), aisladas o asociadas a las de otros sistemas, como el urinario y el digestivo, no dependen de ninguna causa hormonal. Las malformaciones aisladas pueden consistir en ausencia total de trompas y útero y atresia de vagina (síndrome de Rokitansky), atresia de útero o de vagina, útero doble o bicorne e himen imperforado. El único síndrome para el que se han descrito mutaciones monogénicas es el síndrome pie-mano-genital asociado a anomalías en el gen HOXA13 (OMIM 142959)5, con efecto dominante y variable sobre el desarrollo genitourinario.
Lectura rápida
Actualmente está bien establecido que la etiología de un número importante de ADS son mutaciones en genes necesarios para la diferenciación sexual normal. La demostración de la mutación o mutaciones establece el diagnóstico etiológico definitivo y permite los estudios familiares, el consejo genético y el diagnóstico prenatal.
La clasificación actual de las ADS que se acompañan de cariotipo 46,XY se presenta en la tabla 3. El primer apartado incluye las anomalías de diferenciación de la gónada masculina, el testículo, en el segundo se presentan las anomalías en la síntesis o en la acción de los andrógenos, en el tercero las anomalías de síntesis o de acción del factor inhibidor de los conductos de Müller (MIF) y en el cuarto se clasifican síndromes malformativos aislados o complejos. En la tabla 4 se resumen las ADS de causa monogénica o cromosómica conocida con sus características fenotípicas y bioquímicas que pueden ser revisadas junto con el tratamiento en Audí et al2.
Anomalías de la diferenciación sexual 46,XY por anomalías del desarrollo gonadal (testículo)Disgenesia gonadal completa o parcial 46,XYDentro de las anomalías de la diferenciación gonadal con cariotipo 46,XY, la falta completa de diferenciación testicular constituye la disgenesia gonadal pura completa, llamada también síndrome de Swyer, mientras que las formas parciales o incompletas reciben también el nombre de ADS 46,XY disgenético.
- —
Genes DMRT1 y DMRT2 (OMIM 602424)5. Los pacientes 46,XY con monosomía en la región 9p del cromosoma 9, en la región crítica 9p24.3 que contiene los genes DMRT1 y DMRT2, presentan falta de desarrollo testicular, síndrome al que pueden asociarse dismorfia craneofacial y retraso mental. También en el sexo genético femenino 46,XX, la haploinsuficiencia de 9p puede dar lugar a disgenesia gonadal aunque ésta no se presenta en todos los casos12,13.
- —
Gen WNT4 (OMIM 603490)5. Se ha descrito un efecto de dosis para el gen WNT4, localizado en el cromosoma 1p35. Se emitió la hipótesis de que WNT4 suprimiría el desarrollo de las células de Leydig en el ovario; en efecto, ya en el año 198414 se había descrito que la duplicación de 1p provocaba una ADS en un paciente 46,XY, habiendo sido confirmado recientemente que la duplicación del fragmento 1p31-1p35 provoca una ADS 46,XY disgenético8,15,16.
- —
Gen DAX-1 (OMIM 300018)5. La duplicación del gen DAX1 impide la normal diferenciación testicular a pesar de un cariotipo 46,XY y el gen SRY normal17.
- —
Gen SRY (OMIM 480000)5. Se han descrito diversas alteraciones en la región seudoautosómica del cromosoma Y o en el gen SRY: deleciones y translocaciones de parte de la región seudoautosómica del brazo corto del cromosoma Y sobre el cromosoma X o sobre un autosoma, con pérdida de material genético y diversas mutaciones en el gen SRY18,19.
- —
Gen WT-1 (OMIM 607102)5. Se han descrito disgenesias testiculares 46,XY en pacientes con deleciones en 11p13. Se trata del síndrome de Denys-Drash (tumor de Wilms, aniridia, retraso mental y disgenesia gonadal 46,XY o complejo WAGR) y del síndrome de Frasier (cuando no incluye tumor de Wilms), en los que existen mutaciones en el gen WT-1 (Wilms tumor-1), localizado en 11p13. Se han descrito algunos casos de deleciones de 11p13 que asocian disgenesia testicular y gonadoblastoma a la aniridia y el retraso mental, pero sin nefropatía ni tumor de Wilms. Las mutaciones en este gen tienen un efecto autosómico dominante20. En algunos casos de síndrome de Frasier que asocia nefropatía congénita, sin tumor de Wilms, y disgenesia testicular 46,XY no se detectan mutaciones en el gen WT-1; sin embargo, se ha demostrado que los pacientes con síndrome de Frasier presentan mutaciones en el punto de corte y empalme del intrón 9 del gen WT-1, por lo que se produciría una isoforma más larga de la proteína y existirían 2 isoformas, una de ellas anómala21. El estudio sistemático del gen WT-1 en 15 niños con hipospadias pero con función endocrina testicular normal y sin nefropatía ni tumor de Wilms ha permitido demostrar la existencia de 3 mutaciones distintas en 3 de estos niños22.
- —
Gen SOX-9 (OMIM 114290)5. Se había descrito un locus SRA-1 (del inglés sex reversal autosomal locus) en la región cromosómica 17q24.3-25.1 (entre los locus para los genes de GH y tirosincinasa) en los pacientes 46,XY con displasia camptomélica y ADS completo o incompleto por disgenesia gonadal, habiéndose demostrado que el gen mutado era SOX-9 (responsable de la displasia ósea y de la falta de diferenciación testicular)23. El estudio de un paciente con cariotipo 46,XX con virilización24 sugiere que ésta es debida a una duplicación de un fragmento de uno de los cromosomas 17 que contiene SOX-9.
- —
Gen SF-1 o NR5A1 (OMIM 184757)5. El receptor nuclear denominado SF-1 (del inglés steroidogenic factor 1) tiene un papel fundamental en la ontogenia de las suprarrenales, las gónadas y las células gonadotropas. Se describió un paciente con cariotipo 46,XY que presentaba una ADS disgenética con déficit total de respuesta de la T a la estimulación, presencia de derivados de los conductos de Müller y una insuficiencia suprarrenal de presentación neonatal que había sido catalogada de hiperplasia suprarrenal lipoidea25. Recientemente se ha ido demostrando que las mutaciones en el gen SF-1 podrían ser relativamente frecuentes en los pacientes 46,XY con ADS disgenética y que no siempre asocian la insuficiencia suprarrenal; además bastaría un alelo afectado para manisfestarse una ADS 46,XY26.
- —
Gen CBX2 (OMIM 602770)5. El gen CBX2 (localizado en 17q25) acaba de ser incorporado a la lista de genes reguladores de la diferenciación gonadal en los 46,XY ya que se ha descrito una primera paciente 46,XY con diferenciación ovárica y genital interna y externa normales, portadora de 2 mutaciones inactivadoras en CBX227.
- —
Otros genes candidatos y síndromes malformativos asociados a disgenesia gonadal 46,XY. Recientemente se ha descrito el primer paciente 46,XY con una mutación en homocigosis del gen DHH (desert hedgehog) (OMIM 605423)5 que presenta disgenesia gonadal (un testículo disgenético en un lado y una gónada fibrosa en el otro, genitales internos y externos femeninos) asociada a polineuropatía. Se especula que este gen intervendría en la diferenciación testicular.
Las deleciones a nivel Xq13.3 provocan un síndrome que asocia retraso mental, betatalasemia y ADS 46,XY: el gen candidato sería ATRX (OMIM 300032)5. Otro gen en el cromosoma X (ARX, OMIM 300382)5 provoca otro síndrome que asocia lisencefalia, epilepsia y ADS 46,XY. Finalmente, el gen TSPYL1 (OMIM 604714)5 es responsable del síndrome SIDDT con muerte súbita infantil y ADS 46,XY.
Quimera ovotesticular 46,XYYa se ha mencionado en apartados anteriores que los quimerismos ovotesticulares (presencia simultánea en una o ambas gónadas de tejidos testicular y ovárico con diferenciación normal) pueden comportar cariotipos en mosaico o 46,XX o 46,XY, siendo el más frecuente el 46,XX. Su denominación clásica había sido “hermafroditismo verdadero”.
Regresión testicularCuando, a pesar de un cariotipo 46,XY y de una diferenciación genital masculina normal no existen datos anatómicos ni hormonales de la existencia de testes se admite que éstos se diferenciaron y fueron funcionales hasta una cierta etapa de la vida fetal. La delimitación de la cronología de la regresión testicular puede deducirse de la cronología de la diferenciación genital masculina.
Lectura rápida
Para ello es necesario utilizar técnicas de biología molecular. Las mutaciones detectadas pueden ir desde la deleción total del gen, que a veces incluye material genético contiguo, pasando por deleciones parciales (desde varios exones hasta una sola base) e inserciones hasta cambios puntuales de un solo nucleótido que puede provocar desde una parada en la pauta de lectura hasta una proteína con estructura anómala y pérdida de función.
En el testículo fetal, la función de las células de Leydig puede ser anómala, aisladamente, por varias causas, la mayoría de las cuales han sido bien caracterizadas hasta su nivel molecular. Las diversas causas conocidas son: secreción de LH fetal anómala, aplasia o hipoplasia de las células de Leydig, defecto de la biosíntesis del colesterol y déficit enzimáticos de la esteroidogénesis testicular (tabla 3). Las anomalías en la acción de los andrógenos comportan dos posibles niveles: el déficit de 5α-reductasa y las anomalías del receptor de andrógenos; a ellos se añaden una posible iatrogenia de origen materno y el efecto de contaminantes ambientales (tabla 3).
Existe una sola publicación que refiere un caso de ADS 46,XY caracterizado por disminución de la secreción endógena de T, LH elevada y respuesta testicular normal a la estimulación con hCG28. Una anomalía similar fue descrita en un paciente con hipogonadismo y aplasia de células de Leydig que respondían a la estimulación con hCG, habiéndose demostrado la existencia de una mutación puntual en la subunidad β de la LH29.
Mutaciones en el gen LHCGR (OMIM 152790)5 (aplasia o hipoplasia de las células de Leydig)El síndrome consiste en una ADS 46,XY con falta completa de virilización de los genitales externos, en el que el cariotipo 46,XY se acompaña de la presencia de testes criptorquídicos, pequeños, con células intersticiales indiferenciadas que no secretan T y no responden a la estimulación con hCG; no existen derivados de los conductos de Müller, demostrando que la secreción de MIF por las células de Sertoli fetales ha sido normal; la secreción de LH es elevada, mientras que la de FSH es normal. Los testes de estos pacientes presentan células intersticiales con aspecto fibroblástico, que no responden a la LH en cuanto a secreción de T, y se ha demostrado que estas células carecen o tienen un número disminuido de receptores para la LH30.
Se publicaron posteriormente casos con grados variables de afectación de la diferenciación de las células intersticiales, algunos de ellos familiares. Se han descrito casos “parciales”, en los que la denominación correcta sería de “hipoplasia” de células de Leydig o de resistencia parcial de las células de Leydig a la LH.
El gen fue clonado y localizado en el locus 2p16-21 del cromosoma 2. En él se ha descrito, en casos tanto esporádicos como familiares, la existencia de mutaciones con efecto recesivo31.
Déficit enzimáticos en la biosíntesis de testosteronaLos 5 primeros déficit de la esteroidogénesis (StAR, colesterol-desmolasa, 3β hidroxiesteroide-deshidrogenasa, P-450-óxido-reductasa y 17α-hidroxilasa/17, 20-desmolasa) afectan tanto a la suprarrenal como al testículo (y en el sexo femenino al ovario), por lo que habrá déficit conjunto de esteroides suprarrenales y testiculares. En cambio, la última enzima que interviene antes de llegar a la testosterona (17β-hidroxiesteroide-deshidrogenasa o 17-ceto-reductasa) se expresa principalmente en gónadas, por lo que no habrá afectación suprarrenal.
Los genes codificadores de cada una de estas proteínas han sido clonados, su localización autosómica determinada y se describen las mutaciones o anomalías moleculares responsables de la falta o déficit de síntesis de la enzima correspondiente o de la síntesis de una proteína anómala e inactiva (tabla 4).
- —
Mutaciones en gen 7-deshidrocolesterolreductasa (síndrome de Smith-Lemli-Opitz) (OMIM 602858)5. El síndrome de Smith-Lemli-Opitz (SLO) estaba catalogado como síndrome polimalformativo que presentaba múltiples malformaciones entre las cuales se hallaban malformaciones genitales variables en los genotipos 46,XY (asocia malformaciones craneofaciales, retraso mental profundo, retraso del crecimiento, sindactilia, posiblemente también insuficiencia suprarrenal e insuficiencia gonadal). El síndrome está provocado por un déficit en la enzima 7-deshidrocolesterol-reductasa que cataliza la última etapa en la biosíntesis del colesterol, el gen está localizado en el cromosoma 11q13 y se han descrito hasta 66 alelos mutantes de DHCR7 entre 342 alelos estudiados en pacientes con el síndrome de SLO 32.
- —
Mutaciones en genes StAR y colesteroldesmolasa (CYP11A1). La primera enzima de la cadena de esteroidogénesis es la colesteroldesmolasa o 20,22-desmolasa. Es una proteína citocrómica P-450scc (del inglés sidechain-cleavage) localizada en las mitocondrias. Su déficit da lugar a otro de biosíntesis de todos los esteroides suprarrenales y gonadales. Su frecuencia es muy baja, pero más elevada en la población japonesa.
El estudio molecular de casos diagnosticados por la exploración clínica y hormonal no había permitido demostrar la existencia de ninguna mutación en el gen que codifica P-450scc (CYP11A1 en el cromosoma 15) hasta que se demostraron en 2 casos con este diagnóstico sendas mutaciones en el gen que codifica una proteína mitocondrial denominada StAR (del inglés steroidogenic acute regulatory protein) (OMIM 600617)5 regulada por la LH en el testículo y por la ACTH en la corteza suprarrenal. Se han descrito hasta 15 diferentes mutaciones en el gen StAR33.
Más recientemente se han logrado detectar mutaciones en el gen CYP11A1 (OMIM 118485)5 que codifica la proteína P-450scc responsable de la actividad colesterol-desmolasa34.
- —
Mutaciones en 3β-hidroxiesteroide-deshidrogenasa (gen HSD3B2). El déficit enzimático grave provoca una hiperplasia suprarrenal congénita y, en el sexo masculino, una ADS 46,XY (tablas 2-4). La primera mutación descrita en 2 pacientes confirmó que se trataba de una patología del gen de la 3β-HSD de tipo II (OMIM 201810)5. Se han descrito hasta la actualidad 34 mutaciones distintas en un total de 56 pacientes pertenecientes a 44 familias35.
- —
Mutaciones en P450 óxido-reductasa (gen POR). El déficit de P450 óxido-reductasa (gen POR, OMIM 124015)5 afecta a todas las actividades enzimáticas microsomales que necesitan esta flavoproteína donadora de electrones. Entre las enzimas de la esteroidogénesis afecta las actividades de CYP17 y de CYP21, por lo tanto provoca un déficit combinado de 21-hidroxilasa y de 17α-hidroxilasa/17,20-desmolasa. En el sexo genético y gonadal masculino provoca una hiperplasia suprarrenal congénita y un déficit de síntesis de T y por lo tanto una ADS 46,XY9,10 (tablas 2-4).
- —
Mutaciones en 17α-hidroxilasa (gen CYP17) y 17,20-desmolasa o liasa (gen CYP17). Las 2 actividades enzimáticas (17α-hidroxilación y 17,20-desmolasa) son realizadas por una sola proteína citocrómica del retículo endoplásmico (P-450c17) codificada por un gen (CYP17) (OMIM 202110)5 en el cromosoma 10q24-25 que contiene 8 exones. El déficit puede manifestarse de forma heterogénea, de modo que puede predominar el déficit de la actividad 17α-hidroxilasa sobre el de la 17,20-desmolasa, o viceversa. Hasta la actualidad se han descrito más de 120 casos36.
El estudio molecular de los casos diagnosticados de ambos tipos de déficit ha revelado que en los dos existen mutaciones en el gen CYP17 (tabla 4) (OMIM 202110)5. La afectación de la actividad 17,20-desmolasa representaría una forma menos grave del déficit que cuando está afectada la actividad 17α-hidroxilasa.
- —
Mutaciones en 17-cetorreductasa (gen HSD17B3). Es el único déficit enzimático testicular que no afecta a la esteroidogénesis suprarrenal (tablas 2 y 4). El estudio molecular de este déficit enzimático permite la descripción de mutaciones en el gen HSD17B3 (OMIM 605573)5,37,38. Algunas mutaciones se repiten en distintas familias y otras mutaciones de novo se hacen recurrentes.

La acción androgénica ocupa la última etapa en la cadena de pasos necesarios para la diferenciación genital masculina. Existen 2 patologías génicas del mecanismo de acción de los andrógenos que provocan una virilización nula o insuficiente de un feto con sexo genético y gonadal masculino: el déficit de 5α-reductasa y la insensibilidad o resistencia a la acción de los andrógenos, cuyas bases moleculares han sido caracterizadas.
La acción androgénica también puede ser contrarrestada por iatrogenia materna y por contaminantes ambientales.
- —
Déficit de 5α-reductasa (gen SRD5A2). En el déficit de 5α-reductasa se han descrito hasta 29 mutaciones en 11 homocigotos y una elevada proporción de heterocigotos compuestos en el gen para la isoenzima de tipo II (SRD5A2) (expresada en próstata y genitales externos), localizada en el brazo corto del cromosoma 2 (2p23) (OMIM 607306)5,39. En pacientes procedentes de la península Ibérica hemos detectado homocigotos para la mutación Q126R así como heterocigotos compuestos para las mutaciones G115D, Q126R, R171S, N193S, A207D y R246W40; en un paciente de origen chino se ha detectado una mutación puntual R227Q, sólo descrita en Asia, así como una deleción puntual no descrita en otros pacientes41.
- —
Insensibilidad a los andrógenos (gen AR). Se trata, probablemente, de la causa más frecuente de ADS 46,XY, aunque su diagnóstico etiológico a menudo no llegue a realizarse o se realice de forma incompleta. El gen del receptor de los andrógenos (AR) está localizado en la región Xq11-12 (OMIM 313700)5. Actualmente se han detectado más de 400 mutaciones distintas y se estableció una base de datos de las mutaciones en el gen del receptor de andrógenos que se actualiza regularmente (http://www.mcgill.ca/androgendb/). Es interesante señalar que no existe un estrecho paralelismo entre el genotipo y el fenotipo en las mutaciones de los exones 2 a 8, pudiendo la misma mutación en la misma familia dar lugar a un síndrome completo o incompleto. En una serie de 59 pacientes (46 con resistencia completa y 13 parcial) estudiada por nosotros hemos detectado 57 mutaciones distintas, de las cuales 34 no habían sido descritas42. Estas mutaciones incluyen cambios puntuales, microdeleciones e inserciones y se distribuyen a lo largo de todo el gen, siendo el exón 1 el que mayor número de mutaciones presenta42.
Actualmente es recomendable el diagnóstico molecular de los casos de resistencia total o parcial a los andrógenos. Ello es importante, además, para poder establecer un estudio familiar que permita la detección de las mujeres portadoras del cromosoma X anómalo y, en caso necesario, hacer posible el diagnóstico prenatal.
ADS 46,XY por anomalías de la síntesis o de la acción del factor inhibidor de los conductos de Müller (MIF o AMH) (ADS 46,XY interno)- —
Déficit de hormona antimülleriana (mutaciones gen AMH) (OMIM 600957)5. Dentro de la clasificación de los diferentes tipos de ADS 46,XY, la llamada ADS 46,XY interno, “varones con hernia uterina inguinal” u “hombre con útero” ocupa un lugar especial por su rareza. La clonación y el conocimiento de la secuencia del gen que codifica el MIF (OMIM 600957)5, localizado en el cromosoma 19-p13.3-13.2, han permitido el estudio de posibles mutaciones responsables del síndrome. Se han descrito mutaciones puntuales presentes en homocigosis, así como la existencia de heterocigotos compuestos43.
- —
Resistencia a la hormona antimülleriana (gen AMHR) (OMIM 600956)5. El receptor del MIF (gen AMHR2) pertenece a la familia de receptores de tipo 2 de las proteínas relacionadas con el factor de transformación del crecimiento beta (TGF-β) y precisa para la transducción de señal la presencia del receptor de tipo 1. El gen AMHR2 ha sido clonado y se localiza en el cromosoma 12q13 (OMIM 600956)5. En aproximadamente los dos tercios de los pacientes con mutaciones en el gen AMHR2 existe una deleción de 27 pares de bases en el exón 10, por lo menos en un alelo44.
Laura Audí Parera. Unidad de Investigación en Endocrinología Pediátrica. Servicio de Pediatría e Institut de Recerca Hospital Vall d'Hebron. CIBER de Enfermedades Raras, Instituto de Salud Carlos III (CIBERER). Hospital Vall d'Hebron y Universidad Autónoma de Barcelona. Barcelona. España.
Ricardo Gracia Bouthelier. Servicio de Endocrinología Pediátrica. CIBERER (CIBER de Enfermedades Raras, Instituto de Salud Carlos III); Hospital Infantil La Paz y Universidad Autónoma de Madrid. Madrid.
Luis Castaño González. Unidad de Investigación. Endocrinología Pediátrica. CIBERER (CIBER de Enfermedades Raras, Instituto de Salud Carlos III). Hospital de Cruces y Universidad del País Vasco. Baracaldo.
Antonio Carrascosa Lezcano. Unidad de Investigación en Endocrinología Pediátrica. Servicio de Pediatría y Institut de Recerca Hospital Vall d'Hebron. CIBERER (CIBER de Enfermedades Raras, Instituto de Salud Carlos III). Hospital Vall d'Hebron y Universidad Autónoma de Barcelona. Barcelona.
Jesús Barreiro Conde. Unidad de Endocrinología Pediátrica, Crecimiento y Adolescencia. Hospital Clínico Universitario de Santiago de Compostela. Santiago de Compostela.
José Antonio Bermúdez de la Vega. Servicio de Endocrinología Pediátrica. Hospital Virgen de la Macarena. Sevilla.
Antonio Gutiérrez Macías. Servicio de Endocrinología Pediátrica. Hospital Virgen de la Arrixaca. Murcia.

