





The literature on inducers in epilepsy and bipolar disorder is seriously contaminated by false negative findings. This is part I of a comprehensive review on antiepileptic drug (AED) inducers using both mechanistic pharmacological and evidence-based medicine to provide practical recommendations to neurologists and psychiatrists concerning how to control for them. Carbamazepine, phenobarbital and phenytoin, are clinically relevant AED inducers; correction factors were calculated for studied induced drugs. These correction factors are rough simplifications for orienting clinicians, since there is great variability in the population regarding inductive effects. As new information is published, the correction factors may need to be modified. Some of the correction factors are so high that the drugs (e.g., bupropion, quetiapine or lurasidone) should not co-prescribed with potent inducers. Clobazam, eslicarbazepine, felbamate, lamotrigine, oxcarbazepine, rufinamide, topiramate, vigabatrin and valproic acid are grouped as mild inducers which may (i) be inducers only in high doses; (ii) frequently combine with inhibitory properties; and (iii) take months to reach maximum effects or de-induction, definitively longer than the potent inducers. Potent inducers, definitively, and mild inducers, possibly, have relevant effects in the endogenous metabolism of (i) sexual hormones, (ii) vitamin D, (iii) thyroid hormones, (iv) lipid metabolism and (v) folic acid.
La literatura en epilepsia y el trastorno bipolar en los inductores está contaminada por falsos negativos. Esta es la primera parte de una revisión exhaustiva de los fármacos antiepilépticas (FAE) con propiedades inductoras que usa los mecanismos farmacológicos y la medicina basada en la evidencia para aportar recomendaciones prácticas a neurólogos y psiquiatras sobre el modo de controlar sus efectos. La carbamazepina, el fenobarbital y la fenitoina son potentes inductores con efectos clínicos relevantes. Se calculan sus factores de corrección para los fármacos que han sido estudiados. Estos factores de corrección son una simplificación para orientar a los clínicos, ya que existe entre las personas una gran variedad en la intensidad de los efectos inductores. A medida que se publique nueva información, deberán modificarse los factores de corrección. Algunos de estos factores son tan elevados que algunos fármacos (el bupropion, la quetiapina o la lurasidona) no deberán administrarse conjuntamente con los inductores potentes. El clobazam, la eslicarbazepina, el felbamato, la lamotrigina, la oxcarbazepina, el rufinamide, el topiramato, la vigabatrina y el ácido valproico son inductores leves que pueden: a) ser inductores sólo en dosis elevadas; b) combinar a menudo efectos inhibidores, y c) emplear meses en alcanzar sus efectos inductores máximos o su desaparición, e indudablemente necesitan más tiempo que los inductores potentes. Claramente, los inductores potentes, y posiblemente los inductores débiles, tienen efectos relevantes en el metabolismo endógeno de a) las hormonas sexuales, b) la vitamina D, c) las hormonas tiroideas, d) el metabolismo lipídico, y e) el ácido fólico.
In a two-part editorial in which Part I1 focuses on epilepsy and Part II2 focuses on bipolar disorder, the author proposed that the neuropsychopharmacology literature on drug–drug interactions (DDIs) with drug metabolic inducers is seriously contaminated by false negative findings. Inducers’ effects are systematically denied or at least undervalued, and the available published literature systematically deemphasizes their clinical relevance. Moreover, this pattern of denial occurs both in epilepsy1 and bipolar disorder2 literature, where inducers increase the metabolism of many drugs metabolized by the Cytochrome P450 (CYP) and/or Uridine Diphosphate Glucuronosyltransferase (UGT) enzymes. This pattern of denial may be defined as systematic since it contaminates different pharmacological categories: (1) CYP3A4 drugs, (2) non-metabolized drugs, and (3) mild inducer drugs.1,2 Moreover, it contaminates the “narrative” of both epilepsy1 and bipolar disorder2 pharmacological treatments which are characterized by polypharmacy, including the use of potent inducers such as carbamazepine or the more recently introduced mild inducers, such as oxcarbazepine. Historically, denial has usually occurred first in the antiepileptic drug (AED) literature1 and then is repeated in the literature on bipolar disorder.2
This review article attempts to provide a comprehensive review of the AED inducer literature to establish the current status of the field in order to provide practical recommendations to clinicians (Part I). Part II reviews the pharmacological mechanisms behind induction and provides further information to interpret AED DDIs in the real world which frequently include other pharmacological mechanisms besides induction.
Part I includes sections on (1) the literature search (for Parts I and II); (2) the limitations of using a practical approach in providing guidelines for clinicians on inducer DDIs; (3) potent inducers, including subsections on the 3 major potent inducers, carbamazepine, phenobarbital and phenytoin, a brief subsection on primidone, and these potent inducers’ effects on major drug classes; (4) mild inducers, with subsections on clobazam, eslicarbazepine, felbamate, lamotrigine, oxcarbazepine, rufinamide, topiramate, vigabatrin and valproic acid (VPA); and (5) a description of AED inducer effects, which go beyond DDIs and include effects on the metabolism of endogenous compounds.
Literature searchThe author has been conducting PubMed searches in this area for more than 15 years in the process of publishing (1) review articles in this area,3–9 (2) studies of the effects of AED inducers in psychiatry,10–19 and (3) a book with practical guidelines for each AED and/or mood stabilizing agent; the book has more than 1000 different references.20 All of these were published in the context of trying to develop a comprehensive model of personalized prescription in psychiatry.21–26 In that sense, adding or discontinuing an inducer is equivalent to decreasing or increasing the dose of the substrate, which is considered a form of personalized dosing.21 Recommending against using a powerful inducer in the context of using a substrate (e.g., phenytoin in a patient taking quetiapine) is considered a form of personalized drug selection.21
The comprehensive searches for individual AEDs with inductive properties conducted in April 2011 were updated for each AED in December 2013 for this article. A PubMed search for eslicarbazepine pharmacokinetics was also conducted since the author had not completed prior searches on this compound before writing this article.
Limitations of a practical approach in providing guidelines for cliniciansThis review targets clinicians for the purpose of resolving the practical problems regularly encountered by psychiatrists and neurologists who frequently manage patients taking polypharmacy regimens. The author works as a consultant in Kentucky's public mental health system and his daily experience is that psychiatrists and neurologists are unaware that AED inducers exert a major influence on the dosing of other drugs; they have limited understanding of how to correct for these effects. This is a review for practicing clinicians, but it may not completely satisfy readers with a scientific orientation. Two major groups of scientists work in the field of psychopharmacology: the basic scientists who focus on pharmacological mechanisms and those who take a statistical approach, the so-called evidence-based medicine approach (EBM).27
Mechanistic approachPharmacologists working in a laboratory may argue28 with the practical classification of potent and mild inducers used in this review article. Smith et al.28 would point out that all AED inducers, independent of their potency, use the same mechanism for induction; from the pharmacological point of view, potency may be defined by the affinity of the inducer to the induced enzyme. That is obviously correct from the pharmacological point of view, but our ability to extrapolate from this understanding of the pharmacological mechanism behind induction applications for the clinical environment is rather limited. It is insufficient for producing a simplified classification for clinicians fundamentally interested in managing their patients. In that sense, the review of our limited understanding of the pharmacological mechanism behind induction is left for Part II of this article, which can be read by clinicians with further interest. As many specific DDIs have not been systematically studied and no information is available to orient clinical decisions, the author must frequently, in his own clinical practice, extrapolate from other drugs with a similar metabolic profile by following mechanistic pharmacokinetic principles. However, the author has learned to be extremely careful when extrapolating from other pharmacological DDIs, as the literature is systematically biased against the inductive effects of AEDs and is frequently contaminated with incorrect statements that get repeated from one article to the next. The best examples of incorrect statements on the effects of inducers refer to topiramate among AEDs and paliperidone among drugs used in bipolar disorder.
Many articles remind us that topiramate is mainly eliminated unchanged in the urine, but in truth it is partly metabolized (the figure usually quoted is 20%) by CYPs.29 As far as the author knows, the specific CYPs have never been studied. Unfortunately, the literature does not stress that metabolism under normal conditions is not the same as metabolism under induction. Although topiramate metabolism under induction has not been well studied, it is clear that topiramate clearance increases two-fold when taking carbamazepine or phenytoin, which requires doubling the dose.29 Therefore, when powerful inducers are prescribed, a larger proportion of topiramate is metabolized, probably around 40%. The paliperidone prescribing describes a decrease of 37% paliperidone area under the curve (AUC) after taking 400mg/day of carbamazepine for unspecified period. That period recently has been recently described to be 3 weeks30 which is not sufficient to reach maximum carbamazepine induction. For years, based on his experience with risperidone,10,11,13 the author31 had hypothesized that risperidone's main metabolite, marketed as paliperidone which was not supposed to be metabolized according to the marketer, may be like topiramate, quite susceptible to induction. Recently, Yasui-Furakori et al.32 have demonstrated that the author's hypothesis was correct; 600mg/day of carbamazepine for 2–4 weeks was associated with an average reduction in plasma paliperidone concentrations to one-third. This will require multiplying the paliperidone dose by 3 in these patients. It is possible that an even higher paliperidone correction factor will be needed in patients taking higher carbamazepine doses or for longer periods of time.
EBM approachScientists with a strict EBM approach will never be satisfied with the recommendations provided in this review article because many of them are based on case reports or on extrapolations based on what we know about the pharmacokinetic mechanisms of drugs with similar metabolism. Neglecting the value of extrapolation can have deleterious consequences. Based in extrapolative thinking that oxcarbazepine may be a less potent inducer that carbamazepine, the author hypothesized that oxcarbazepine has inductive effects on lamotrigine and subsequently detected in real life two cases of initial Stevens Johnson syndrome,12 preventing the progression of a potentially lethal adverse drug reaction (ADR).
EBM approaches are naturally limited in the literature on inducers since, as the author has described,1,2 the current literature is contaminated by a systematic bias denying the clinical relevance of AED inducers. It is very easy to design a randomized clinical trial (RCT) using AED inducers which will produce negative results; one only has to prescribe the inducers using doses or durations below those needed to detect induction; a negative trial will result and one can conclude that inducer A has no effect on substrate B.1,2 Unfortunately, few RCTs have been designed by pharmaceutical companies to study DI with inducers in clinically relevant conditions in order to provide recommendations for clinicians.
This author could then conduct a discussion of the type errors in statistics, which is relevant for the EBM approach, but he prefers to acknowledge that we do not live in an ideal world. In an ideal world, one would like to follow EBM principles by conducting hundreds of RCTs. They would provide recommendations to clinicians on appropriate co-prescription of AED inducers in situations of polypharmacy. Conducting hundreds of RCTs would be quite expensive and require massive effort. In the real world, which includes the absence of extensive funding in this area, the author has been involved in using already-collected samples to explore inductive effects. The most successful outcome has resulted from his valproic acid (VPA) studies on clozapine metabolism. In the first study,14 a sophisticated mathematical model demonstrated that VPA may be an inducer of clozapine metabolism by using a convenience sample comprised of a total of 415 plasma clozapine samples collected for other purposes. These samples were obtained for therapeutic drug monitoring (TDM) in 83 patients sent by their clinicians for clinical purposes and for previously published DI studies in 172 patients.14 Unfortunately, the same type of mathematical model was unable to demonstrate VPA inductive effects on olanzapine in a sample of convenience including a total of 360 plasma olanzapine samples.15 The samples were obtained by combining TDM from 116 patients and DI studies from 47 patients.15 The lack of induction of VPA on olanzapine metabolism was somewhat surprising since olanzapine and clozapine metabolism are rather similar. To detect VPA inductive effects, a more sophisticated design was required; a much smaller olanzapine study (18 patients, 3 samples per patient, a total of 46 olanzapine samples) with a prospective design demonstrated VPA inductive effects on olanzapine metabolism.16 Unfortunately, this prospective study raised more questions as well by indicating that VPA may also competitively inhibit olanzapine metabolism and that VPA duration may be important in determining the net effects (induction versus inhibition).17 As a matter of fact, it is possible that four weeks of VPA treatment in this prospective study was not long enough to reach maximal induction of olanzapine metabolism.
Although the prior paragraph stresses the need of higher levels of evidence in DDI, a completely developed EBM approach may not resolve these questions for all patients. Well-controlled studies tend to focus on average patients but, in the clinical world, many patients, called outliers by statisticians, do not follow average patterns and are not well represented by the means. The author has no knowledge of patients who are completely resistant to inducers and who show no clinically relevant induction of potent inducers using doses and durations expected to cause it, but he would not be surprised if they exist. He has extensive experience with the relatively rare patients (probably <1%) who appear to have very powerful responses to inducers. A few of them are briefly described in Table 1, since the literature rarely acknowledges their existence.*
List of patients with extreme sensitivity to inductive effects.
| Case 1: Caucasian ♂ inpatient with Schizophrenia and late-onset epilepsya |
| A) Extreme sensitivity to CYP3A4 induction |
| -Carbamazepine: 1500–2000mg/dayb was needed in the past to reach therapeutic serum concentrations. |
| -Quetiapine |
| On phenytoin and VPA, the quetiapine C/D ratio was 10 times lower than expected. |
| On VPA, the quetiapine C/D ratio was 2–6 times lower than expected. |
| -Diazepamc |
| On phenytoin, serum concentrations were undetected despite taking 30mg/day. |
| On VPA, diazepam clearance after 30mg IM was ≥3 times higher than expected. |
| B) Possible VPA auto-induction when taking concentrate, but not present on divalproex sodium |
| On concentrate, 5250mg/dayd was needed to reach therapeutic concentrations. |
| On divalproex sodium, 2000mg/daye was enough for therapeutic concentrations. |
| C) Metabolism of clozapine and olanzapine (CYP1A2 drugs) was normal for a male smoker.f |
| Case 2: African-American ♂ inpatient with schizoaffective disorderg |
| A) Extreme clozapine induction by VPA (divalproex sodium) |
| Smoking (20 cigarettes/day): ≥650mg/day was needed for reaching serum concentrations >350ng/ml.h |
| Smoking and VPA: ∼1200mg/day was needed for reaching serum concentrations >350ng/ml.i |
| During smoking induction, the patient demonstrated a high metabolic capacity for clozapine. |
| Addition of VPA induction led to the highest clozapine metabolic capacity the author has seen. |
| Case 3: Caucasian ♂ inpatient with schizoaffective disorderj |
| A) Extreme sensitivity to CYP3A4 induction |
| -Carbamazepine: Up to 2800mg/dayb was needed to reach therapeutic serum concentrations; this is partly explained by deposits in fat tissue and high volume of distribution.k |
| -Risperidone |
| On carbamazepine, approximately 4 times higher than normal metabolic capacity. |
| On divalproex sodium, risperidone metabolism was normal. |
| -Paliperidone: On carbamazepine, high capacity to metabolize paliperidone. |
| B) Extreme olanzapine induction by VPA (divalproex sodium) and omeprazole |
| -Olanzapine: On divalproex sodium and omeprazole, 1.5–2 times higher than normal metabolism. |
| Case 4: Caucasian ♂ inpatient with bipolar disorderl |
| A) Possible VPA auto-induction when taking divalproex sodium |
| -ischarged on 4000mg/daym |
| Case 5: ♂ inpatient with frontal and temporal lobe epilepsy and psychosisn |
| A) Extreme sensitivity to UGT induction during phenytoin and phenobarbital treatment |
| -Lamotrigine: 2.6 times higher than the recommended dosage was needed.o |
| -Lorazepam: Doses >20mg/day were tolerated with no sedation. |
| Case 6: ♂ inpatient with tuberous sclerosis and kidney tumorsp |
| A) Increased VPA metabolic capacity when taking phenytoin and VPA |
| -Divalproex sodium: Dose increased >10,000mg/day to reach therapeutic serum concentrations.q |
| -Phenytoin: No changes in phenytoin metabolism and no need for changes in dosage. |
Usual carbamazepine doses to reach therapeutic concentrations: 800–1200mg/d. Maximum recommended dose is 1600mg/day.124
Diazepam is metabolized primarily by CYP2C19; CYP3A4 is an auxiliary enzyme. In this patient CYP3A4 was probably the primary metabolic enzyme for diazepam.
At the time of highest carbamazepine dose, BMI was 40 with weight of 191kg. The high dose is partly explained by obesity.127
Patient needed 1600mg/day to get therapeutic serum lamotrigine concentrations. The maximum recommended dose is 600mg/day.
Followed for 4 years (ages 44–48 until he died). He initially had angiomyolipomas in both kidneys. In the second year, a growing right kidney mass led to possible diagnosis of renal carcinoma and nephrectomy pathology that suggested angiomyolipoma. In the third year, brain metastasis became evident.
In the beginning when the patient had bilateral kidney tumors, he needed around 5000mg/day of divalproex sodium to get therapeutic concentrations with VPA C/D ratios of 0.010–0.018. After nephrectomy and obvious metastatic renal cancer was present, he needed 10,500mg/day of divalproex sodium to get therapeutic concentrations with VPA C/D ratios of 0.005–0.009.
In reviewing the literature, the author has tried to combine the mechanistic pharmacological and EBM approaches in an attempt to obtain the best recommendations for clinicians at this time. The reinterpretation of vigabatrin and clobazam potential for DDIs may be the best examples of this combined thinking by the author. Both drugs were used in Europe for many years and were not considered inducers. Their introduction in the USA mandated Food and Drug Administration (FDA) requirements such as in vitro studies of their potential for DDIs, which demonstrated that both drugs are mild inducers. This has led the author1 to review the old RCT that, in effect, suggested that these drugs were mild inducers, but the old articles ignored or even rejected the possibility that these two drugs were inducers. Vigabatrin and clobazam teach the lesson that the literature in this area is quite unreliable. Based on the limitations of old literature, one can easily prognosticate that in 5 years this review article may be obsolete and need extensive modifications, including dosing modifications.
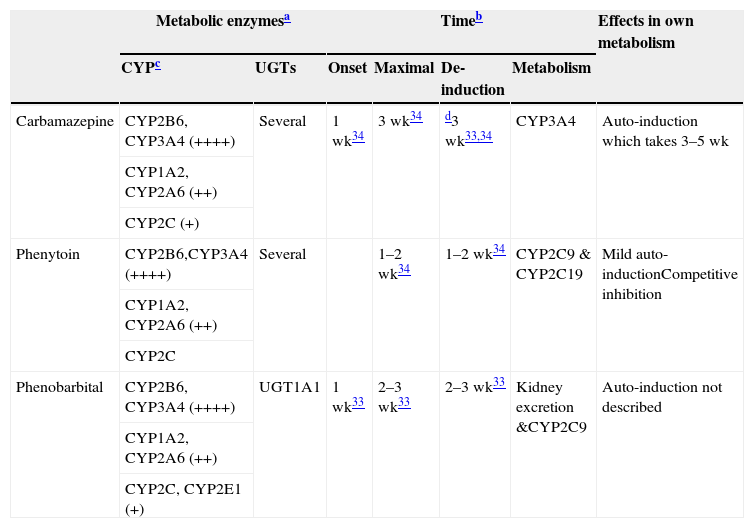
Potent inducersThere is general agreement in the literature33,34 that three AEDs, carbamazepine, phenobarbital and phenytoin, are clinically relevant inducers. In reality, all barbiturates are believed to be inducers, although this review only briefly mentions another one of them, primidone. Table 233–36 provides a summary for clinicians of the common characteristics of these potent inducers. Clinicians interested in better understanding the various CYPs and UGTs and the relevance of their induction are encouraged to look at Table 3. Some information indicates that it is possible that phenytoin may be a more promiscuous and potent inducer than carbamazepine, since it has been reported that adding phenytoin may require doubling the dosage of carbamazepine,37 which indicates that phenytoin pushes the induction beyond carbamazepine's own self-induction. As phenobarbital is currently much less frequently used in developed countries, there is very limited information on its inductive properties in clinical samples.
Characteristics of potent inducers.
| Metabolic enzymesa | Timeb | Effects in own metabolism | |||||
|---|---|---|---|---|---|---|---|
| CYPc | UGTs | Onset | Maximal | De-induction | Metabolism | ||
| Carbamazepine | CYP2B6, CYP3A4 (++++) | Several | 1 wk34 | 3 wk34 | d3 wk33,34 | CYP3A4 | Auto-induction which takes 3–5 wk |
| CYP1A2, CYP2A6 (++) | |||||||
| CYP2C (+) | |||||||
| Phenytoin | CYP2B6,CYP3A4 (++++) | Several | 1–2 wk34 | 1–2 wk34 | CYP2C9 & CYP2C19 | Mild auto-inductionCompetitive inhibition | |
| CYP1A2, CYP2A6 (++) | |||||||
| CYP2C | |||||||
| Phenobarbital | CYP2B6, CYP3A4 (++++) | UGT1A1 | 1 wk33 | 2–3 wk33 | 2–3 wk33 | Kidney excretion &CYP2C9 | Auto-induction not described |
| CYP1A2, CYP2A6 (++) | |||||||
| CYP2C, CYP2E1 (+) | |||||||
++++: massive induction; ++: moderate induction; +: mild induction. wk: weeks.
These are approximated times provided by review articles.29,30 Readers may need to be aware that few studies have been conducted to verify these times.
Not all CYPs have the same ability to be induced by potent inducers. More details are provided in Part II, Table 1. It is believed that potent inducers have massive effects (++++) on CYP2B6 and CYP3A4. On the other hand, potent inducers have only mild to moderate effects on the CYP2C subfamily which includes CYP2C8, CYP2C9 and CYP2C19.35 Although the literature is not specific on this point, the author believes that CYP1A2 may be induced intermediately between the potent effects on CYP2B6 and CYP3A4 and mild effects on the CYP2C subfamily and described as moderate (++). There is limited information on CYP2A6 suggesting potential for moderate induction (++), but clinicians need to be aware that few drugs are metabolized by CYP2A6; this is the main metabolic pathway for nicotine. There is limited information on CYP2E1 which may have mild potential for induction, but clinicians need to be aware that few drugs are metabolized by CYP2E1, although it is a minor metabolic pathway for alcohol and some antiepileptic drugs.
The loss of induction may take longer for CYP1A2 substrates than for CYP3A substrates (respective induction half-lives were 105 and 70h, or 4.4 days and 2.9 days.36
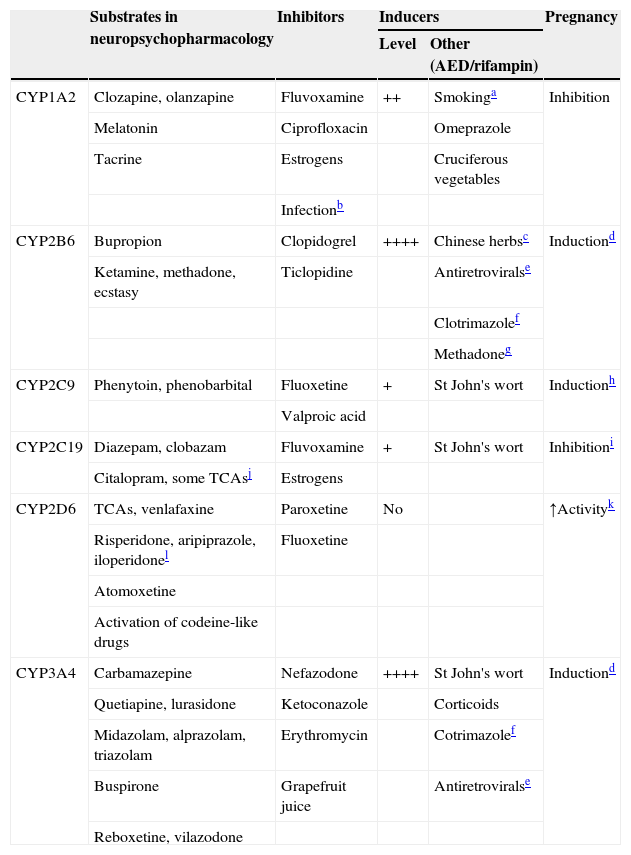
CYPs involved in drug metabolism.
| Substrates in neuropsychopharmacology | Inhibitors | Inducers | Pregnancy | ||
|---|---|---|---|---|---|
| Level | Other (AED/rifampin) | ||||
| CYP1A2 | Clozapine, olanzapine | Fluvoxamine | ++ | Smokinga | Inhibition |
| Melatonin | Ciprofloxacin | Omeprazole | |||
| Tacrine | Estrogens | Cruciferous vegetables | |||
| Infectionb | |||||
| CYP2B6 | Bupropion | Clopidogrel | ++++ | Chinese herbsc | Inductiond |
| Ketamine, methadone, ecstasy | Ticlopidine | Antiretroviralse | |||
| Clotrimazolef | |||||
| Methadoneg | |||||
| CYP2C9 | Phenytoin, phenobarbital | Fluoxetine | + | St John's wort | Inductionh |
| Valproic acid | |||||
| CYP2C19 | Diazepam, clobazam | Fluvoxamine | + | St John's wort | Inhibitioni |
| Citalopram, some TCAsj | Estrogens | ||||
| CYP2D6 | TCAs, venlafaxine | Paroxetine | No | ↑Activityk | |
| Risperidone, aripiprazole, iloperidonel | Fluoxetine | ||||
| Atomoxetine | |||||
| Activation of codeine-like drugs | |||||
| CYP3A4 | Carbamazepine | Nefazodone | ++++ | St John's wort | Inductiond |
| Quetiapine, lurasidone | Ketoconazole | Corticoids | |||
| Midazolam, alprazolam, triazolam | Erythromycin | Cotrimazolef | |||
| Buspirone | Grapefruit juice | Antiretroviralse | |||
| Reboxetine, vilazodone | |||||
++++: massive induction; ++: moderate induction; +: mild induction. AED: antiepileptic drug.
Polycyclic aromatic hydrocarbons in smoke have inductive effects. These compounds also found on chargrilled food and coffee from roasted coffee beans, which can also have inductive effects.128
Respiratory infections, other serious infections, such as pyelonephritis or appendicitis or even major inflammations can inhibit CYP1A2 because the cytokines released inhibit CYP1A2.
Sodium ferulate was an inducer of bupropion metabolism in a study. It is the sodium salt of ferulic acid, which is widely distributed in herbs and Chinese formulas such as Ligusticum, Chuanxiong and Chaihu–Shugan–San.129 Another inducer is baicalin, a flavone glucuronide extracted from the medical plant Radix scutellariae, which is present in fruits, vegetables, and beverages derived from plants (tea, red wine), and in a wide range of herbal medicines including Huang-Lian-Jie-Du-Tang, hangeshashinto, San-Huang-Xie-Xin-Tang, Da-Chai-Hu-Tang, and Xiao-Chai-Hu-Tang.130
Pregnancy definitively induces CYP2B6 and CYP3A4.131 According to an in vitro study, CYP2B6 and CYP3A4 are induced by both estrogen and progesterone. Progesterone also induces CYP3A5.
In vitro studies indicate that methadone may induce its own metabolism and this may be mediated not only by CYP2B6 but also by CYP3A4.137
It is believed that CYP2C9 increases during pregnancy because phenytoin clearance increases. It cannot be ruled out that mechanisms other than CYP2C9 induction may explain changes in phenytoin clearance during pregnancy. Estradiol increases the activity of CYP2C9 without affecting expression by unknown mechanisms.138
Estrogens are thought to be competitive inhibitors of CYP2C19, but a recent study suggested that they may inhibit CYP2C19 expression.139
CYP2C19 is the main enzyme to demethylate amitriptyline, clomipramine and imipramine. Their metabolites then are further metabolized by hydroxylation, mainly by CYP2D6.
It is not well understood why CYP2D6 activity may increase in pregnancy, since it is believed that CYP2D6 cannot be induced. A recent study suggested that pregnancy may remove a suppressor of CYP2D6 expression.140
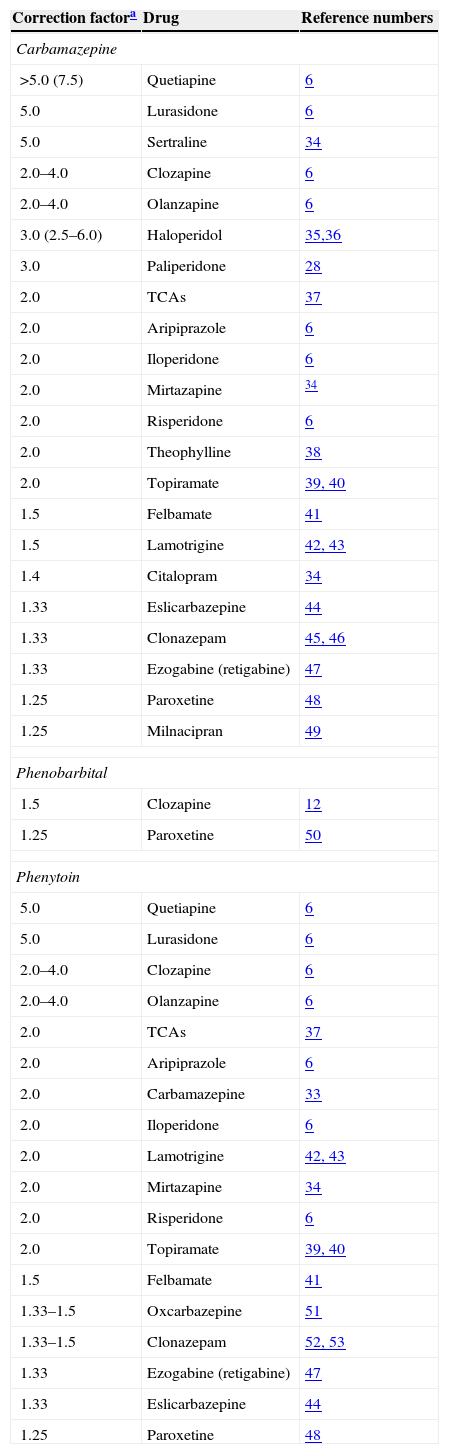
Table 4 provides correction factors calculated by the author using the available literature.6,14,32,38–57 Adding a potent inducer requires increasing the dose of the substrate. For example, a correction factor of 2.0 indicates that the substrate dose should be doubled. If the patient is taking an inducer and a substrate, and the inducer is discontinued to maintain the same plasma concentration of the substrate, one should use the inverse of the correction factor 1/2.0=0.5, or halve the dose. Table 4 provides recommendations based on the limited information available; as more articles are published; these correction factors will need to be recalculated. References are provided in case the reader wants to update them. Remember that correction factors are average corrections for average patients; thus, an individual may have a metabolic capacity higher or lower than average. Sometimes TDM can be used to make more precise corrections in a specific individual.6,25
Correction factors for potent inducers.
| Correction factora | Drug | Reference numbers |
|---|---|---|
| Carbamazepine | ||
| >5.0 (7.5) | Quetiapine | 6 |
| 5.0 | Lurasidone | 6 |
| 5.0 | Sertraline | 34 |
| 2.0–4.0 | Clozapine | 6 |
| 2.0–4.0 | Olanzapine | 6 |
| 3.0 (2.5–6.0) | Haloperidol | 35,36 |
| 3.0 | Paliperidone | 28 |
| 2.0 | TCAs | 37 |
| 2.0 | Aripiprazole | 6 |
| 2.0 | Iloperidone | 6 |
| 2.0 | Mirtazapine | 34 |
| 2.0 | Risperidone | 6 |
| 2.0 | Theophylline | 38 |
| 2.0 | Topiramate | 39, 40 |
| 1.5 | Felbamate | 41 |
| 1.5 | Lamotrigine | 42, 43 |
| 1.4 | Citalopram | 34 |
| 1.33 | Eslicarbazepine | 44 |
| 1.33 | Clonazepam | 45, 46 |
| 1.33 | Ezogabine (retigabine) | 47 |
| 1.25 | Paroxetine | 48 |
| 1.25 | Milnacipran | 49 |
| Phenobarbital | ||
| 1.5 | Clozapine | 12 |
| 1.25 | Paroxetine | 50 |
| Phenytoin | ||
| 5.0 | Quetiapine | 6 |
| 5.0 | Lurasidone | 6 |
| 2.0–4.0 | Clozapine | 6 |
| 2.0–4.0 | Olanzapine | 6 |
| 2.0 | TCAs | 37 |
| 2.0 | Aripiprazole | 6 |
| 2.0 | Carbamazepine | 33 |
| 2.0 | Iloperidone | 6 |
| 2.0 | Lamotrigine | 42, 43 |
| 2.0 | Mirtazapine | 34 |
| 2.0 | Risperidone | 6 |
| 2.0 | Topiramate | 39, 40 |
| 1.5 | Felbamate | 41 |
| 1.33–1.5 | Oxcarbazepine | 51 |
| 1.33–1.5 | Clonazepam | 52, 53 |
| 1.33 | Ezogabine (retigabine) | 47 |
| 1.33 | Eslicarbazepine | 44 |
| 1.25 | Paroxetine | 48 |
Bupropion's correction factor for carbamazepine was 10.0, calculated by the author from the limited information available.49
Carbamazepine is a classic AED that is approved in the US for partial onset epilepsy, bipolar disorder and trigeminal neuralgia.20 Carbamazepine is mainly metabolized by CYP3A (with a minor contribution from CYP2C8) to its active metabolite, 10,11-epoxide, which may account for 40% of carbamazepine metabolism, but the proportion is even greater in patients with induced CYP3A4 activity. The epoxide is subsequently transformed into the inactive diol by an epoxide hydroxylase. Other pathways include aromatic hydroxylation (25%), possibly by CYP1A2, and glucuronidation of the carbamoyl side chain by UGTs, presumably primarily by UGT2B7.20
Carbamazepine induces its own metabolism (Table 2), increasing it by three times.20 When starting carbamazepine, levels may not be in steady state for the first 3–5 weeks due to the progressive increase in auto-induction. Table 4 provides correction factors for several drugs during carbamazepine treatment.
PhenobarbitalPhenobarbital is a classic AED that is approved in the US for generalized and partial onset epilepsy and for sedation.20 Phenobarbital is eliminated unchanged in urine (20–50%) and metabolized to parahydroxyphenobarbital and phenobarbital N-glucoside. CYP2C9 plays a major role in the formation of parahydroxyphenobarbital. Other minor metabolizing enzymes are CYP2C19 and CYP2E1.20
The literature does not indicate that phenobarbital causes auto-induction (Table 2). Knowing its inductive profile of being a CYP2C9 inducer, it is possible that self-induction occurs but it is probably modest and not easy to detect due to phenobarbital's extraordinarily long half-life (several days). Table 4 provides correction factors only for clozapine and paroxetine since there is no information for other drugs. If there is need, the recommendation is to use correction factors for carbamazepine and/or phenytoin as rough approximations.
The literature suggests that phenobarbital's AED inductive effects are mild (and probably not clinically relevant, <1/3 decrease in levels) for several AEDs including felbamate; lacosamide, levetiracetam, pregabalin and rufinamide.20
PhenytoinPhenytoin is a classic AED that is approved in the US for generalized and partial onset epilepsy and prevention of seizures secondary to surgery or head trauma.20 Phenytoin is extensively para-hydroxylated by CYP2C9 while CYP2C19 may be the second most important enzyme for this step. Other less important enzymes may be CYP2C8 and CYP3A.20 Phenytoin is a mild inducer of its own metabolism, according to a study in volunteers58 which is compatible with in vitro studies showing only mild to moderate inductive effects on the CYP2C subfamily.35 More importantly than auto-induction, clinicians need to be very aware that phenytoin can inhibit its metabolism by saturating CYP2C9 and CYP2C19. Phenytoin can be described pharmacologically as having a narrow therapeutic window and following nonlinear pharmacokinetics; its kinetics are dose-dependent and capacity-limited. In the experience of the author, dosing beyond the recommended upper range of the therapeutic window of >20mcg/ml yields the saturation of the enzymes and the increase of half-life, which will require the complete discontinuation of phenytoin for at least 2–3 days until normal metabolism recovers to levels <20mcg/ml and closely followed TDM. In situations of high plasma phenytoin concentration (close to 20mcg/ml), adding any CYP2C inhibitor or any drug that competes with CYP2C9 and/or CYP219 may be accompanied by dramatic concentration increases that probably would not occur if phenytoin concentrations were considerably lower (e.g., 10mcg/ml). Table 4 provides correction factors for several drugs during phenytoin treatment.
PrimidonePrimidone is a classic AED that is approved in the US for generalized and partial onset epilepsy. It is rarely used in epileptic patients resistant to other treatments and those with essential tremors.20 Up to one quarter of primidone is eliminated unchanged in urine and the remainder is metabolized by the CYP2C isoenzymes to two active metabolites: phenobarbital and phenylethylmalonamide. As primidone is transformed into phenobarbital, one should consider it as powerful an inducer as phenobarbital.20Table 4 does not provides correction factors for primidone; if there is need, the author would suggest using phenytoin's correction factors as an approximation.
Effect of potent inducers on major drug classesThis subsection comments on AEDs, psychiatric and medical drugs. AEDs are too heterogeneous in their metabolism to be briefly reviewed as a group, but Table 4 provides data on correction factors for several of them when potent inducing AEDs are administered. Three psychiatric drug classes (antipsychotics, antidepressants and benzodiazepines) and non-neuropsychopharmacological drugs metabolized by CYP3A4 are reviewed briefly in this section.
When clinicians want to consider adding a potent inducer in a patient taking an antipsychotic, they should consider the risk of induction and review the most current literature. A recent review of second-generation antipsychotics6 indicated that antipsychotics can be divided into three groups: those needing massive dose increases, those needing minimal or no increases, and those in an intermediate group needing moderate to significant dose increases. Lurasidone and quetiapine need massive increases in dosing (see Table 4). In that 2012 review article, amisulpride, paliperidone and ziprasidone were classified as needing minimal or no increases but as previously indicated, the recent study from Yasui-Furakori et al.32 demonstrated that paliperidone should be eliminated from this group of antipsychotics needing minimal or no increases after adding potent AED inducers and include in the group requiring moderated increased. Thus, the intermediate second-generation antipsychotics are those dependent for their metabolism on CYP1A2 and UGTs (clozapine and olanzapine), those partly dependent on CYP3A4 (aripiprazole, iloperidone and risperidone), and probably paliperidone (see Table 4). Currently, the author has no information to indicate whether potent inducers influence asenapine metabolism or not.6 There is very limited information on correction factors for first-generation antipsychotics (see Table 4 for haloperidol).
The information of the effects of potent inducers on tricyclic antidepressants (TCAs) is limited. Tertiary amines such as amitriptyline, clomipramine and imipramine are demethylated by CYY2C19 and, to a lesser extent, CYP1A2, CYP2C9 and CYP3A4; all of these CYPs can be induced.10 Secondary amines such as nortriptyline and desimipramine are hydroxylated by CYP2D6 and possibly other CYPs.10 Thus, if a patient is taking a TCA and potent inducers of TCAs, TDM should be used. Table 4 provides an approximate correction factor of 2.0, but the range, based on review,41 is 1.4–2.5. This indicates that it is better to use TCA TDM when trying to correct for the effect of a potent inducer in a patient taking a TCA. There is very limited information on the effects of potent inducers on the metabolism of second-generation antidepressants.8,38,53,59Table 4 provides correction factors for citalopram, milnacipran, mirtazapine, sertraline and paroxetine in patients taking carbamazepine; for paroxetine in patients taking phenobarbital; and for citalopram and paroxetine in patients taking phenytoin. Bupropion is included as Footnote a in Table 4 because 10.0 is the correction factor calculated by the author from the limited information available.8 Bupropion is mainly metabolized by CYP2B6, which is very sensitive to induction; therefore, bupropion should be avoided in any patient taking potent inducers since the pharmacy may not want to dispense bupropion in doses 10 times higher than usual. Reboxetine and vilazodone, which are mainly metabolized by CYP3A4, may be particularly sensitive to induction. According to data from antipsychotics dependent on CYP3A4, they may require a correction factor of at least 5.8
Benzodiazepines are heterogeneous in their metabolism but three major groups exist: those metabolized by (1) CYP3A4, (2) CYP2C19 and CYP3A4, and (3) UGTs. CYP3A4 accounts for 93%60 of triazolam's metabolism, 92%60 of midazolam's, and 75%60 of alprazolam's. These three benzodiazepines are likely to be strongly induced by potent inducers and they should be avoided in patients taking potent inducers. Clonazepam is partly metabolized by CYP3A4; Table 4 provides correction factors for carbamazepine and phenytoin. Lorazepam and oxazepam are not metabolized by CYPs but by UGTs.61 No studies exist, but potent inducers are likely to have clinically relevant effects on lorazepam and oxazepam. Similarly, there are no good studies of the effects of potent inducers on diazepam and clobazam, which are metabolized by CYP3A4 and CYP2C19. The limited clobazam data62,63 indicates that, although carbamazepine is likely to be an inducer, phenytoin may induce CYP3A4 but also compete with CYP2C19 metabolism.6
All three potent AED inducers are potent CYP3A4 inducers. Many non-neuropsychopharmacological drugs are extensively metabolized by CYP3A4, including many calcium channel blockers, statins, immunosuppressants and estrogen-containing oral contraceptives. Clinicians combining carbamazepine, phenytoin, or phenobarbital with any drug from any of these drug classes must pay attention to the lack of efficacy associated with decreased plasma concentrations and consider increasing doses of these substrates or switching to similar drugs not metabolized by CYP3A4. If the drugs are mainly metabolized by CYP3A4, the correction factor will be ≥5. This is based on the correction factor of quetiapine (Table 4), which has been estimated to be 85% metabolized by CYP3A4.64 The case of antiretroviral agents is particularly complex. All three potent AED inducers should be avoided in patients taking protease inhibitors or non-nucleoside reverse transcriptase inhibitors.65
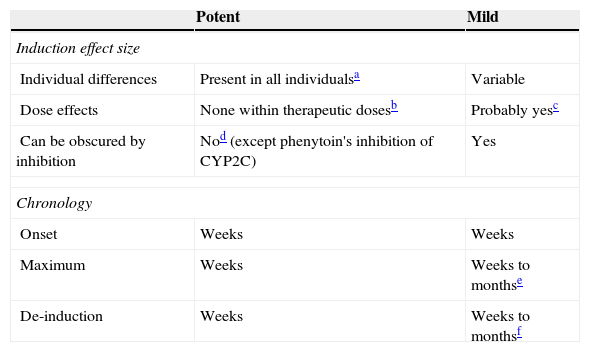
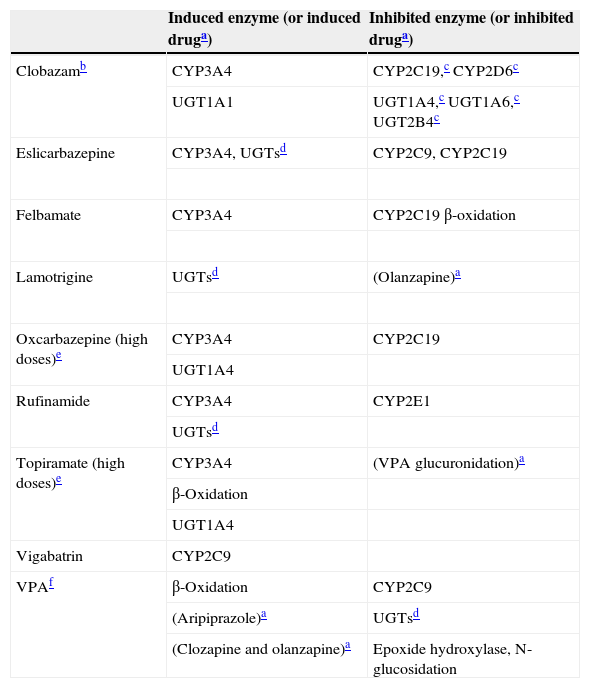
Mild inducersThis article groups clobazam, eslicarbazepine, felbamate, lamotrigine, oxcarbazepine, rufinamide, topiramate, vigabatrin and VPA as mild inducers. As far as the author knows, no other author has grouped them together in the same way, nor has contrasted them to the powerful inducers (Table 5). Thus, a summary of their inductive enzymes includes their inhibitory properties (Table 6) to help the reader understand that these AEDs have a very complex DDI profile.
Mild inducers: comparison to potent inducers.
| Potent | Mild | |
|---|---|---|
| Induction effect size | ||
| Individual differences | Present in all individualsa | Variable |
| Dose effects | None within therapeutic dosesb | Probably yesc |
| Can be obscured by inhibition | Nod (except phenytoin's inhibition of CYP2C) | Yes |
| Chronology | ||
| Onset | Weeks | Weeks |
| Maximum | Weeks | Weeks to monthse |
| De-induction | Weeks | Weeks to monthsf |
Although it has not been systematically studied, it is generally accepted that potent inducers tend to maximally induce all patients as long as they are given doses beyond those causing maximal induction.
It is also usually believed that a therapeutic dose for epilepsy should cause maximal induction in most patients. Therefore, further increased doses beyond therapeutic doses may not cause more induction. Similarly, giving another potent inducer to a person taking usual doses of one potent inducer may not make a difference.
Mild inducers: their inductive and inhibitory properties.
| Induced enzyme (or induced druga) | Inhibited enzyme (or inhibited druga) | |
|---|---|---|
| Clobazamb | CYP3A4 | CYP2C19,c CYP2D6c |
| UGT1A1 | UGT1A4,c UGT1A6,c UGT2B4c | |
| Eslicarbazepine | CYP3A4, UGTsd | CYP2C9, CYP2C19 |
| Felbamate | CYP3A4 | CYP2C19 β-oxidation |
| Lamotrigine | UGTsd | (Olanzapine)a |
| Oxcarbazepine (high doses)e | CYP3A4 | CYP2C19 |
| UGT1A4 | ||
| Rufinamide | CYP3A4 | CYP2E1 |
| UGTsd | ||
| Topiramate (high doses)e | CYP3A4 | (VPA glucuronidation)a |
| β-Oxidation | ||
| UGT1A4 | ||
| Vigabatrin | CYP2C9 | |
| VPAf | β-Oxidation | CYP2C9 |
| (Aripiprazole)a | UGTsd | |
| (Clozapine and olanzapine)a | Epoxide hydroxylase, N-glucosidation |
VPA: valproic acid.
For drugs in parentheses, the enzyme behind the induction or inhibition is not definitively established.
In the 1970s clobazam was first approved in Australia and then in France for anxiety and epilepsy. In 2011 it was approved in the US for the adjunctive treatment of seizures associated with Lennox–Gastaut syndrome.9 Clobazam's major metabolic pathway involves N-demethylation, primarily by CYP3A4, and to a lesser extent by CYP2C19 and CYP2B6. N-desmethylclobazam is an active metabolite extensively metabolized, mainly by CYP2C19. N-desmethylclobazam and its metabolites comprise approximately 94% of the total drug-related components in urine.9
In vitro studies indicate62,63 that (1) N-desmethylclobazam is a weak inhibitor of CYP2D6, CYP2C9, UGT1A4, UGT1A6 and UGT2B4; and (2) clobazam and N-desmethylclobazam induced CYP3A4 activity in a concentration-dependent manner. It is surprising that clobazam is a mild CYP3A4 inducer because clobazam is a benzodiazepine, and benzodiazepines are not thought to be inducers.
Therefore, clobazam auto-induces its own metabolism (since it is metabolized by CYP3A4 and is an inducer of CYP3A4); however, by carefully reviewing an old study,66 this author1 has concluded that auto-induction may not start until the third week of treatment. As in vitro studies indicate, the inductive effects depend on both clobazam and N-desmethylclobazam and are concentration-dependent. It is important to know that N-desmethylclobazam requires several weeks to reach steady state,9 and it is likely that clobazam maximal induction may take months until the effects of steady state N-desmethylclobazam are completely developed.
Clobazam is definitively a midazolam inducer63 which is not surprising since midazolam is mainly (92%)60 metabolized by CYP3A4. The US prescribing information offers conflicting interpretations of clobazam induction's clinical relevance, since it states that dosage adjustment of drugs that are primarily metabolized by CYP3A4 is not needed but then states that adding clobazam may be associated with the loss of oral contraceptive efficacy,62 which could only be explained by CYP3A4 induction.
The literature provides conflicting information on clobazam's inductive effects of carbamazepine, which is mainly metabolized by CYP3A4, but at the same time induces its own metabolism and can be partly inhibited by N-desmethylclobazam.1 Similarly, the clobazam information on the possibility of UGT induction is conflicting.1 Clobazam's US prescribing information62 reported that population pharmacokinetic studies during the clobazam RTCs indicated no effects on the metabolism of valproate and lamotrigine while some,67 but not all, TDM studies1 indicate that clobazam may be a mild inducer of lamotrigine metabolism. A TDM study indicated that clobazam may mildly reduce serum levetiracetam concentrations.68
EslicarbazepineEslicarbazepine acetate is a second-generation AED launched onto the European market in 2009 for adjunctive treatment in adult subjects of partial-onset epilepsy,69 but not yet available in the US. After absorption, eslicarbazepine acetate is rapidly and extensively (95%) hydrolyzed in the first-pass metabolism at the liver and gut to eslicarbazepine (also known as S-licarbazepine or S-MHD). Another 5% is oxidized to oxcarbazepine and R-licarbazepine (or R-MHD). Metabolites eliminated in the urine include 2/3 of the total dose as S-MHD and 1/3 as glucuronide conjugates probably mediated by UGT1A1.70 Eslicarbazepine acetate is mainly a pro-drug of S-MHD.69
Eslicarbazepine does not self-induce its own metabolism, but appears to be a clinically relevant inducer of CYP3A4, demonstrated by increasing the clearance of simvastatin by approximately 271 and of the ethinyl estradiol and levonorgestrel present in oral contraceptives in a dose-dependent manner.72 Eslicarbazepine may also be a weak UGT inducer, slightly increasing (<20%) the clearance of several AEDs including carbamazepine, lamotrigine and topiramate.48 In vitro studies indicate that eslicarbazepine is a moderate CYP2C9 and CYP2C19 inhibitor, which is compatible with increasing phenytoin levels by one-third.48
FelbamateFelbamate is a second-generation AED approved in the US for epilepsy that responds inadequately to alternative treatments and is so severe that a substantial risk of aplastic anemia and/or liver failure is deemed acceptable.20 Approximately 40–60% of felbamate is excreted by the kidneys and is metabolized by hydroxylation (by CYP3A and CYP2E1) and glucuronidation. CYP3A contributes in a greater extent to its metabolism when inducers are prescribed. Felbamate is an inhibitor of CYP2C19 and β-oxidation and an inducer of CYP3A.20 There is very limited information on the relevance of felbamate's inductive effects.
LamotrigineLamotrigine is a second-generation AED approved in the US for several types of epilepsy and for maintenance treatment of bipolar disorder.20 Glucuronidation is the major metabolic pathway, accounting for up to 65–90% of lamotrigine metabolism.61 The main metabolite is the inactive 2-N-glucuronide. Lamotrigine and its metabolites are eliminated in the urine. Reviews suggest that UGT1A4 may metabolize lamotrigine. There is disagreement on the importance of UGT27B as a metabolic enzyme for lamotrigine.20
Lamotrigine is a weak inducer of glucuronidation and of its own metabolism (<20% reduction within 2 weeks). The effect of this auto-induction is not seen in patients already taking more potent inducers.20
Lamotrigine may be associated with small (<25%) reductions in VPA levels.73 There are relatively few well-controlled studies on the effects of lamotrigine on antipsychotic levels.5,6 A mild decrease of quetiapine metabolism was described in a quetiapine TDM study,74 but a more recent TDM study described a decrease of approximately half, which will require doubling the quetiapine dose.75 The best clozapine studies do not show major effects on clozapine levels.5,6 The olanzapine data is more complicated and includes an in vitro study suggesting that high lamotrigine concentrations can inhibit olanzapine metabolism76; clinical studies failed to demonstrate lamotrigine inhibitory effects.5,6 The lack of consideration given to smoking status may explain this; lamotrigine might behave as a mild inhibitor of olanzapine metabolism only in smokers.15 Lamotrigine is not expected to influence second-generation antidepressants.8,38
OxcarbazepineOxcarbazepine is a second-generation AED that is approved in the US for partial-onset epilepsy.20 Some clinicians, based on its similarity with carbamazepine's chemical structure and fewer ADRs, use oxcarbazepine for bipolar disorder and trigeminal neuralgia.
Oxcarbazepine is rapidly reduced by a cytosol arylketone reductase to MHD, also called licarbazepine, which is the clinically relevant metabolite.20 Therefore, oxcarbazepine acetate is a pro-drug of both enantiomers S-MHD (80%) and R-MHD (20%).69 MHD is cleared by glucuronidation and, less so, by oxidation to an inactive metabolite. Renal excretion is the major route for oxcarbazepine excretion (80% of dose) including glucuronides of MHD (40%); unchanged MHD (27%); conjugates of MHD or oxcarbazepine (13%); and, in small proportion, the inactive 10,11-dihydroxymetabolite.20
Oxcarbazepine is not an inducer of its own metabolism, as carbamazepine is.77 Oxcarbazepine is a weak inducer of CYP3A and glucuronidation enzymes and a weak CYP2C19 inhibitor.
The literature generally agrees that its inductive effects are less pronounced than those of carbamazepine.78 There is more disagreement on whether oxcarbazepine's inductive effects are mild and can be ignored, or are clinically relevant. Unfortunately, the literature has not paid attention to Patsalos et al.,79 who proposed that only oxcarbazepine doses ≥1500mg/day may have inductive effects because this may explain the conflicting findings. Oxcarbazepine frequently shows no inductive effects in the low to moderate doses used in controlled studies, but appears to be a mild but clinically relevant inducer in naturalistic studies. In the best published pharmacokinetic study, Andreasen et al.80 compared 17 days of 1200mg/day of oxcarbazepine and 800mg/day of carbamazepine in healthy volunteers; clearance of a CYP3A-dependent quinidine metabolite respectively increased around 90% and 180%. If one assumes that this study accurately reflects what happens to CYP3A4 drugs, doses of these drugs would need to be almost doubled when oxcarbazepine is added versus almost tripling them when carbamazepine is added.
A controlled study using 900–1200mg/day for 5 weeks showed no relevant effects on risperidone or olanzapine metabolism.81 Clinicians should be alert that oxcarbazepine, particularly in high doses, may increase the metabolism of some psychiatric drugs. In fact, a case has been published of a clinically relevant decrease in clomipramine levels after adding oxcarbazepine.82
A controlled study in healthy subjects performed by a pharmaceutical company suggested that oxcarbazepine doses of 1200mg/day do not influence lamotrigine metabolism in patients taking 200mg/day.83 A TDM study using long-term treatment suggested that serum lamotrigine levels may be mildly reduced when oxcarbazepine is co-administered, and lamotrigine doses may need to be increased by approximately 20–30%.47,84 The clinical relevance of this DDI is demonstrated by two cases of lamotrigine-induced oral ulcers (initial signs of Stevens–Johnson syndrome) two months after oxcarbazepine discontinuation.12 Oxcarbazepine reduces levetiracetam and topiramate levels by <30%.78,85 Oxcarbazepine may also be a minor (21% decrease) inducer of carboxylesterases that metabolize rufinamide.86
In summary, high doses of oxcarbazepine can induce CYP3A4, UGT1A4 and possibly other metabolic enzymes. Controlled studies using ≥1200mg/day of oxcarbazepine are needed to establish the magnitude of its inductive effects in the average patient and the correction factor needed to compensate. Naturalistic studies and case reports need to consider the possibility that some individuals may be particularly sensitive to oxcarbazepine inductive effects and demonstrate induction in lower doses. Oxcarbazepine de-induction may take up to 2 months to completely manifest.
RufinamideRufinamide is a second-generation AED that is approved in the US only for use in Lennox–Gastaut syndrome.20 Rufinamide is extensively metabolized (only 2–4% is excreted unchanged in urine and feces). It is not metabolized by CYPs but by carboxylesterases. There are no known active metabolites.20 Rufinamide is a weak inhibitor of CYP2E1, a weak inducer of CYP3A, and possibly a weak inducer of some glucuronidation enzymes.20
The effects of rufinamide on other antiepileptic levels may not be clinically relevant. Small decreases (<15%) appear to occur with carbamazepine and lamotrigine levels.87 Currently, another rufinamide DDI that is considered relevant is that rufinamide is an inducer of oral contraceptives. Rufinamide may have no effects on topiramate and VPA levels.87
Plasma levels of psychiatric drugs metabolized by CYP3A may be mildly reduced when co-administered with rufinamide, due to the induction of CYP3A, but this interaction has not been systematically studied except for triazolam, a CYP3A4 substrate (which may account for 93%60 of triazolam's metabolism). Rufinamide increased triazolam metabolism probably by one-third but this was estimated with a single-dose of triazolam.87 It is difficult to extrapolate to repeated dosing of CYP3A4 substrates in clinical practice, which may be much more influenced by rufinamide.
TopiramateTopiramate is a second-generation AED that is approved in the US for several types of epilepsy and migraine prophylaxis20 and in combination with phentermine for weight loss.
Topiramate is mainly eliminated unchanged in the urine but is partly metabolized by CYP (approximately 20%). The relative importance of CYP metabolism increases when taking a powerful inducer, such as phenytoin or carbamazepine, which doubles topiramate clearance due to an increase in CYP metabolism.29
Topiramate also appears to be a weak inducer of several metabolic enzymes, but induction may be influenced by topiramate doses. After an in vitro study, Nallani et al.88 proposed that in doses ≥400mg/day topiramate inductive properties may have clinical significance for CYP3A substrates. In a cohort of 12 women with epilepsy receiving stable dosages of VPA along with a combination norethindrone, 1mg/ethinyl estradiol, 35-μg tablet, topiramate doses of 200mg/day, 400mg/day, or 800mg/day caused a statistically significant dose-related decrease in the mean ethinyl estradiol AUC by 18–30% at the 200- to 800-mg/day dose level.89 The pharmacokinetics of norethindrone remained unchanged. In contrast, another study found that topiramate doses of 50–200mg/day did not significantly affect the clearance of either ethinyl estradiol or norethindrone.90
Topiramate may have complex effects on VPA since it may serve as an inducer by increasing β-oxidation but can also inhibit VPA glucuronidation. At low VPA doses β-oxidation is the most important VPA metabolic pathway and topiramate may behave as an inducer of VPA metabolism.91 At high VPA doses, glucuronidation is the most important VPA metabolic pathway and topiramate may behave as an inhibitor of VPA metabolism.92
A small controlled study completed by the company with progressive increases in dosages of up to 400mg/day for 2 weeks showed no relevant DDI between lamotrigine and topiramate.93 In a controlled study by independent investigators, topiramate was added to lamotrigine treatment monthly from 100mg/day to up to 800mg/day if tolerated; the study demonstrated that in 4 of 7 patients there was a decrease in lamotrigine metabolism by 40–50%.94 Reimers et al.,95 in a large lamotrigine TDM study controlled for confounding factors, suggested that topiramate may decrease lamotrigine levels, but the topiramate doses were not described.
In summary, high doses of topiramate can induce CYP3A4 and some other enzymes (Table 6). Controlled topiramate studies using ≥400mg/day are needed to establish the magnitude of its inductive effects in the average patient and the correction factor needed to compensate. Naturalistic studies and case reports need to consider the possibility that some individuals may be particularly sensitive to topiramate inductive effects and demonstrate them in lower doses. A complicating factor is that in some situations topiramate may have drug metabolism inhibitory properties.
VigabatrinVigabatrin was first licensed as an antiepileptic agent in the UK and the Republic of Ireland in 1989. By the late 1990s, it had been accepted into mainstream clinical practice in the care of adult and pediatric patients but reports of severe, persistent visual field defects were associated with vigabatrin. Twenty years later, vigabatrin was introduced in the US market for patients with refractory complex partial seizures who have inadequately responded to several alternative treatments and for whom the potential benefits outweigh the risk of vision loss.
According to the US prescribing information,96 vigabatrin is not significantly metabolized and it is eliminated primarily through renal excretion. In a study of isotopes using a single-vigabatrin dose to explore drug clearance, Durham et al.97 found that about 95% of total radioactivity was recovered in the urine, with the parent drug representing most of it (82%); a metabolite accounted for 3–5% of radioactivity and another for 1–2%. There are no studies on whether repeated dosing increases vigabatrin's metabolized fraction or not. Even assuming that <10% of vigabatrin is metabolized, there is no doubt that the fraction of vigabatrin that is metabolized increases remarkably in patients taking inducers, although the increase has not been sufficiently well quantified in the literature.1 Vigabatrin has very rarely been prescribed as monotherapy and most patients studied in the literature were taking several other AEDs; most frequently they were taking powerful inducers such as carbamazepine or phenytoin. Therefore, in the typical vigabatrin patient, metabolism may be clinically relevant and include more than 10% of the dose, due to the presence of inducers.
An in vitro study used to gain approval in the US market demonstrated that vigabatrin is a CYP2C9 inducer,96 which explains some old findings of reduction of plasma phenytoin concentrations by 25% that were not evident until the 5th or 6th week.98 The US prescribing information97 recommends that, although phenytoin dose adjustments are not routinely required, dose adjustments of phenytoin should be considered if clinically indicated. Mild decreases in VPA and phenobarbital levels have also been described.96
VPAVPA is a classic AED that is approved in the US for several types of epilepsy, bipolar disorder and migraine prophylaxis.20 VPA undergoes hepatic metabolism with <5% eliminated unchanged in the urine. Major metabolism occurs by UGTs (40%) and β-oxidation as a fatty acid (30%) with minor CYP-dependent metabolism (including CYP2C9, CYP2C19 and CYP2A6). At low doses, β-oxidation may be the most important pathway, while at therapeutic doses glucuronidation may be more important. Many UGTs appear to be involved in VPA glucuronidation including UGT1A3, UGT1A4, UGT1A6, UGT1A9, and UGT2B7 and the primarily intestinal UGT1A8 and UGT1A10.20
VPA is usually considered an inhibitor of several enzymes (Table 6) including CYP2C9, epoxide hydroxylase, several UGTs and the N-glucosidation pathway of phenobarbital.20 Studies in rats appeared to suggest that VPA auto-induced its own glucuronidation.99 The first suggestion that VPA may auto-induce its own metabolism were from studies focused on other drugs (felbamate100 and lamotrigine101). Exploring their effects on VPA led their authors to question whether VPA can auto-induce its own metabolism in humans.
More recently, information on its inductive properties has been accumulating. VPA induces: (1) its own metabolism by inducing β-oxidation (prospective study)102; (2) CYP3A4 and P-gp gene expression (in vitro study)103; (3) possible UGT1A1 in a patient taking irinotecan (which has an active metabolite SN-38 metabolized by UGT1A1)104; (4) aripiprazole metabolism to a mild degree (prospective study)105; (5) olanzapine metabolism (case series,106 TDM,107 and a prospective DI study)15; (6) clozapine metabolism (case series,108,109 prospective case,18 and statistical model on TDM-DI studies14,19); and (7) vitamin D metabolism in an in vitro study.110 The available information on clozapine-olanzapine metabolism indicates that VPA can be an inducer and/or a competitive inhibitor. Its net effect (predominance of induction or inhibition) may depend on time, VPA dose and the presence/absence of smoking.
Other effects of AED inducers beyond DDIIn an excellent recent review on the inductive properties of AEDs from the point of view of epilepsy, Brodie et al.111 provide a historical perspective, recognizing that the problem has been studied for the last 30 years112 but an awareness of its magnitude for endogenous metabolism has been apparent only in the last 10 years. The prior sections focused on the metabolism of exogenous compounds (called xenobiotics), but potent inducers have other long-term health consequences, since their inductive properties may influence the metabolism of endogenous compounds and homeostasis at several levels including (1) sexual hormones, (2) vitamin D, (3) thyroid hormones, (4) lipid metabolism and (5) possibly folic acid. In the author's experience, probably due to endogenous compensatory mechanisms, these inductive effects of endogenous compounds are much less likely to be detected and to be clinically relevant than those occurring in these compounds are administered as exogenous drugs.
It is obvious that CYP3A4 inducers have powerful inductive effects on sexual hormones administered as medications. Moreover, several mild inducers (clobazam, eslicarbazepine, oxcarbazepine, topiramate and rufinamide) have been approved with warnings about pregnancy risk in women using oral contraceptives as the only contraceptive method. The inductive effects are also observed when sexual hormones are administered for treating hypopituitarism.113 The effect of potent inducers on endogenous female and male sexual hormones appears more limited, although in rare cases they can be associated with diminished potency in men and menstrual disorders in women.114
Vitamin D, as with sexual hormones, is a derivative compound of cholesterol; potent inducers definitively interfere with its metabolism, which is mediated by CYPs, resulting in the potential for osteoporosis.115 Moreover, more recent data suggest that mild inducers, including oxcarbazepine, topiramate and valproic acid, may also cause osteoporosis due to the induction of vitamin D metabolism.110,116
UGTs are fundamental for thyroid hormone metabolism, but potent inducers can interfere with thyroid function117 by increasing thyroid hormone metabolism. Occasionally, oxcarbazepine has also been associated with hypothyroidism.113,117 In the author's experience, thyroid abnormalities are more likely when the endogenous feedback is impaired and frequent when thyroid medication is administered exogenously and the body cannot compensate for the mild thyroid dysfunction caused by AEDs. Although it is not well understood, potent inducers may interfere with the metabolism of cholesterol and other complex lipids and have been associated with hyperlipidemia.118
More controversial is the idea that potent inducers may influence folic acid metabolism, which may contribute to hyperhomocysteinemia, a possible risk factor for atherosclerosis.119,120 Phenytoin is clearly associated with gingival overgrowth;121 phenytoin and phenobarbital can cause macrocytic anemia,122 which has been associated with disturbances in folic acid metabolism. The literature usually reports that phenytoin may be an inhibitor of the folic acid transporter.121,122
In summary, the effect of potent inducers in the endogenous metabolism of: (1) sexual hormones, (2) vitamin D, (3) thyroid hormones, (4) lipid metabolism, and (5) folic acid are established. The possible milder effects of some mild inducers in some of these endogenous pathways are starting to be described in the literature.
ConclusionThe author has proposed that the neuropsychopharmacology literature on DDI in both epilepsy1 and bipolar disorder2 is currently seriously contaminated by false negative findings. This comprehensive review of the AED inducer literature provides practical recommendations to clinicians, while Part II reviews the pharmacological mechanisms behind induction and how future studies can address the current deficiencies in the literature.
Part I acknowledges that even a comprehensive literature review cannot eliminate the limitation of literature biased toward negative findings. In reviewing the literature, the author has tried to combine the mechanistic pharmacological and EBM approaches to produce the best recommendations for clinicians at this time. However, he acknowledges that it is likely that in 5 years this review article may be obsolete and the correction factors provided (Table 4) may need to be extensively modified.
There is general agreement in the literature33,34 that three AEDs, carbamazepine, phenobarbital and phenytoin, are clinically relevant inducers (Table 2); however, all barbiturates, including primidone, should be included as such. Table 4 provides correction factors calculated by the author using the available literature to correct for the inductive effects of carbamazepine, phenobarbital and phenytoin. The author needs to acknowledge that using correction factors is a rough simplification for orienting clinicians, since there is great variability in the population regarding inductive effects.123 The author started using correction factors over a decade ago3 due to the absence of such practical information in published reviews. Some of the correction factors are so high that other drugs should not co-prescribed with potent inducers. Among second-generation antipsychotics that should not be prescribed with potent inducers are lurasidone and quetiapine. Bupropion is the clearest case of a second-generation antidepressant that should not be co-prescribed with potent inducers. Phenytoin, besides being a potent inducer for multiple enzymes, can cause clinically relevant CYP2C9 and CYP2C19 inhibitions by saturation of these enzymes when plasma phenytoin concentrations are >20mcg/ml.
The author acknowledges that grouping clobazam, eslicarbazepine, felbamate, lamotrigine, oxcarbazepine, rufinamide, topiramate, vigabatrin and VPA as mild inducers has never occurred in the literature, but it is justified because they have common elements (Table 5) and, more importantly, may help prescribers to learn more about this neglected issue. All of them tend to have mild inductive effects, frequently combined with inhibitory properties (vigabatrin is the exception; Table 6). They appear to take months for reach maximum effects or de-induction and definitively longer than the potent inducers (Table 5). Five mild inducers, clobazam, eslicarbazepine, felbamate, oxcarbazepine, in high doses (≥1200mg/day), and topiramate in high doses (≥400mg/day), are CYP3A4 inducers and are likely to have clinically relevant effects on drugs highly dependent on CYP3A4 for their metabolism, including oral contraceptives, antipsychotics such as lurasidone and quetiapine, and benzodiazepines such as triazolam, midazolam and alprazolam. VPA's inductive properties on clozapine and olanzapine metabolism require more study, but the author has published information which clearly establishes that it can be clinically relevant in some patients.
Potent inducers, definitively, and mild inducers, possibly, have relevant effects on the endogenous metabolism of: (1) sexual hormones, (2) vitamin D, (3) thyroid hormones, and (4) lipid metabolism.
Conflict of interestNo commercial organizations had any role in the writing of this paper for publication. The author reports no financial relationship with commercial interests in the last 36 months.
The authors acknowledge Lorraine Maw, M.A., and Margaret T. Boden, R.N., M.L.T. at the Mental Health Research Center at Eastern State Hospital, Lexington, KY, USA, who helped in editing the article.
Please cite this article as: de Leon J. Efectos de los inductores antiepilépticos en la neuropsicofarmacología: una cuestión ignorada. Parte I: Resumen para los clínicos de la situación actual. Rev Psiquiatr Salud Ment (Barc). 2015;8:97–115.
Tables 1 and 3 belong in fact to the second part of this article, but are also reproduced here for easing the reading.
articles