





Systemic lupus erythematosus (SLE) is an autoimmune disorder with a genetic basis, and is characterized by the appearance of autoantibodies, the formation and deposition of circulating immune complexes, and chronic inflammation in various organs.
It is of multifactorial origin, and in genetically predisposed individuals, environmental factors and hormonal components play a key role in the immune system of the disease. About 25 genetic loci have been identified, indicating the importance in this pathology. However, the concordance rate for SLE is only 25% among monozygotic twins.
An example of this is the deficiencies of the initial components in the classical serum complement pathway such as C1q, C2 or C4, which, although rare, confer genetic susceptibility for SLE at a rate of 30% in the case of C4 deficiency, and more than 90% for C1q deficiency.
It was also demonstrated that C1q inhibits plasmacytoid dendritic cells (pDCs) in the secretion of interferon-alpha (IFN-α), thus providing a new link between complement deficiency and activation of the IFN pathway.
Therefore, IFN-α is considered to have a central role in the pathogenesis of SLE, with high serum concentrations being found in outbreaks of this disease.
These IFN exert prominent immunoregulatory effects, suggesting that this cytokine is key, not only in the innate immune system, but also in adaptive immune responses.
Taking these facts into account, it can be anticipated that pDCs, the main source of IFN secretion, are involved in this autoimmune disease.
In this review, we will focus on the participation of and IFNs in SLE.
El lupus eritematoso sistémico (LES) es un trastorno autoinmune con base genética, caracterizado por la aparición de autoanticuerpos, formación y depósito de complejos inmunes circulantes e inflamación crónica en varios órganos.
La etiología es multifactorial y, en individuos genéticamente predispuestos, factores medioambientales y componentes hormonales juegan un rol clave en el sistema inmune de esta enfermedad.
Cerca de 25 loci genéticos han sido identificados, indicando la importancia en esta enfermedad; sin embargo, la tasa de concordancia para el LES es de tan solo el 25% entre gemelos monocigotos1,2.
Un ejemplo de ello son las deficiencias de los componentes iniciales en la vía clásica del complemento sérico como el C1q, C2 o C4, que si bien es infrecuente, confieren susceptibilidad genética para el LES en una tasa del 30% en caso de deficiencia del C4 y de más del 90% para una deficiencia del C1q3.
Por otro lado, se demostró que el C1q inhibe a las células dendríticas plasmocitoides (CDP) en la secreción de interferón alfa (IFN-α), proporcionando así un nuevo enlace entre la deficiencia del complemento y la activación de la vía del IFN4.
Por ello, el IFN-α es considerado como un actor central en la patogénesis del LES, encontrándose concentraciones séricas altas en los brotes de esta enfermedad5.
En consecuencia, estos IFN ejercen efectos claves en la fisiopatología del LES, lo que sugiere que esta citoquina no solo posee un efecto a nivel del sistema inmune innato, sino también en las respuestas inmunes adaptativas.
Teniendo en cuenta estos hechos, se puede anticipar que las CDP, fuente principal de secreción de IFN, están involucradas en dicha enfermedad autoinmune.
En esta revisión nos centraremos en la participación de las CDP y del IFN en el LES6,7.
Systemic lupus erythematosus (SLE) is an autoimmune disease whose pathogenesis is multifactorial, caused by genetic and environmental factors and alterations of the immune system.1–3
The incidence of SLE is higher in certain ethnic groups, such as Asians, Latin Americans and African North Americans. The disease follows a chronic course with periods of remission and exacerbation. Almost any organ can be affected; its presentation is heterogeneous, ranging between a mild cutaneous manifestation and manifestations of the renal or central nervous systems that endanger life.4,5
Several genes have been associated with susceptibility to SLE; each of them has a small effect that suggests this link. However, the interactions between the gene and the environment are still subject of research.
Regarding the immune system, B cells have been the first therapeutic targets in SLE; the importance of the innate immune system and, in particular, the pathways involved in the interferon signaling (IFN) are emerging.6 Currently, there are data supporting a key role for plasmacytoid dendritic cells (pDCs), with a number of biological therapeutic agents and small-molecule inhibitors that are in the research phase.
Recent data point to alternative forms of modulation of the pathway of IFN and of the pattern recognition receptors.7,8
Dendritic cellsDendritic cells (DCs) are the most important antigen-presenting cells that activate the virgin T cells and perform functions in the innate responses against infections and jointly between the innate and the adaptive immune systems.
Discovered at the end of the 1970s by Ralph Steinman and Cohn Zanvil,9 these cells have an extraordinary ability for immune responses to foreign antigens, acting as true “sentinels” of the immune system.
It is important to mention that DCs are classified based on different criteria such as: their localization in the body, their maturity stage or their origin.
Classification according to their locationDCs circulate in serum as myeloid or lymphoid precursor cells, representing approximately 1% or peripheral blood mononuclear cells (PBMCs), in non-lymphoid tissues. At the level of the skin are found the epidermal DCs, also known as “Langerhans cells” (LCs),10 while in the dermis there are “dermal DCs”,11 which belong to a broader subpopulation of “interstitial DCs”12 that are present in the majority of organs, such as the liver, the kidneys, the heart and other connective tissues. The “DCs associated with mucosal surfaces” are found in the mucosa of the oral cavity, and of the intestinal and respiratory systems. These populations of DCs which are present in non-lymphoid tissues act as sentinel cells capturing antigens in the barriers to entry into the body. Once the antigens have been captured, the DCs migrate to the lymphoid organs where they carry out the presentation of antigen to the T lymphocytes in order to activate them. Although the term LCs is mainly used to refer to the DCs of the epidermis, this term has been extended to the DCs present in all stratified epithelia.13 In lymphoid tissues, the germinal center, which is the microenvironment that allows the generation of memory B lymphocytes, also contains “follicular DCs” and “germinal center DCs” (GCDCs). The GCDCs are potent antigen-presenting cells to T lymphocytes.14,15
The DCs that have captured antigens and have migrated from the skin, non-lymphoid interstitial tissues and mucosal surfaces to the afferent lymph, are recognized as “afferent lymph DCs”, also called “veiled or veliform cells”.16 These migrating cells represent an intermediate phase between LCs (immature DCs par excellence) and the “interdigitating DCs” (IDCs) into which they are transformed.17 The latter are present mainly in secondary lymphoid organs, they have a high degree of maturation and can initiate immune responses by activation of virgin T lymphocytes. Contrary to what happens with the LCs, “the DCs of the thymus” (thymic DCs) appear to be non-migrating cells, which are generated in the thymus, where they complete their life cycle.18
Classification by their maturity stageDCs have been demonstrated in different works which compared: LCs isolated in fresh (“immature”) and LCs from epidermal suspensions subsequently cultured in vitro (“matured”) revealed their different capabilities of antigenic uptake and processing and of stimulation of T lymphocytes.19 By way of definition, immature DCs are characterized by having high phagocytic and antigenic processing capacity, being located mainly in peripheral regions of the body such as the skin and mucous membranes and having a smaller number of molecules of the major histocompatibility complex (MHC-II) and costimulatory molecules. In contrast, mature CDs are directed to the T zones of the secondary lymphoid organs, where their capacity for the presentation of antigens to the T lymphocytes is reflected, being their phagocytic activity more limited. In addition, they overexpress the MHC-II molecule superficially (and not in the cytoplasm as the immature ones) and costimulatory molecules.20 These definitions are in line with the classical model in which the immature DCs present in the skin and mucous membranes mature during their migration to the lymph nodes, decreasing their capacity of antigen uptake and increasing their ability to stimulate the T lymphocytes.21
Classification according to the originHematopoietic and mesenchymal systemThe DCs of hematopoietic origin are divided in turn into DCs from lymphoid and myeloid progenitors.22,23 When attempting to classify the DCs based on their origin and functionality, we make use of a broad terminology that sometimes can be very complicated and ambiguous. This is the case of meanings such as “myeloid”, “lymphoid”, “conventional”, “plasmacytoid”, “type 1 DC”, “type 2 DC”, “IPDCs” (IFN producing DCs), making it even more complicated due to the different subdivision of DCs that is made when talking about murine and human models, which are the most studied species. Therefore, the most common way to classify the DCs is the one that divides them into conventional (mainly of myeloid origin) and plasmacytoid (primarily of lymphoid origin).24
The pDCs were identified for the first time in paracortical areas of reactive lymph nodes, with morphology similar to that of the plasma cells and with markers common to T lymphocytes and monocytes. They have long membrane projections with phagocytic capacity, widely distributed in the lymphatic tissues, the mucosal epithelium and the parenchyma of the organs.25 It was also observed that these cells produced large amounts of type 1 IFN in response to certain viruses and that under certain culture conditions (IL3, DC40L) they were transformed into cells with stellate morphology and with the ability to activate virgin T lymphocytes. Thus, plasmacytoid T lymphocytes, plasmacytoid monocytes or natural-IFN producing cells turned out to be the same cell type called pDC.26–28
And those of mesenchymal origin, whose basis are follicular DCs, differ from the rest of DCs because they are typical stromal cells of connective tissue and the only thing they share in common with those of hematopoietic origin is the dendritic morphology. In this sense, the follicular DCs are not true antigen presenting cells since they do not express MHC-II molecules and, therefore, they do not present processed antigens to CD4 T lymphocytes. Instead, they present complete antigens in the form of immune complexes to B lymphocytes, helping to their activation or selective maturation and contributing to the formation of germinal centers, plasma cells and memory B lymphocytes.29,30
The dysfunction of apoptosis and its relationship with the DCs leads to the release of autoantigens, which initiate an immune response; therefore, it is thought that it is one of the physiopathological basis of SLE.31 The DCs recognize and process antigens for their presentation to CD4 T lymphocytes, and in animal models it was demonstrated that the loss of self-tolerance leads to hyperactivity of CD4 T cells and, finally, to the development of autoimmune disease. The phagocytosis of the final product of apoptosis leads to maturation of myeloid DCs (MDCs) and the production of proinflammatory cytokines, including IL-6.32 In the presence of IL-6 and other proinflammatory cytokines, the mature MDCs can induce the activation of Th1, Th2 and Th17 cells, while IL-6 does not allow the differentiation of virgin T cells into regulatory T cells (Treg) in SLE.33
The interferon familyThere are 3 types of IFN known today (IFN I, IFN II, and IFN III). Perhaps the most studied and best known is the IFN I, whose main secretory cells are the pDCs. These IFNs I, all with a considerable structural homology, are encoded by genes located on chromosome 9. The most important IFNs I in the defense against viruses are interferon alpha (IFN-α) (which actually contains 13 different closely related proteins) and interferon beta (IFN-β), which is a single protein.34,35
The IFNs I, and particularly IFN-α, have emerged as key pathogenic cytokines in SLE. Their functions are antiviral, antiproliferative and immunomodulatory effects.36 Signaling through IFN I begins after the union with the IFN receptor (IFNAR): a receptor complex consisting of 2 transmembrane proteins, IFNAR1 and IFNAR2, which together with the union with two cytoplasmic tyrosine-kinases, JAK1 and TYK2, allows the phosphorylation of the 2 proteins (STAT and STAT 2); in addition, IRF9 forms altogether the so called “transcription complex factor” ISGF3, which finally stimulates in the nucleus genetic factors for the formation of IFN37 (Fig. 1).
 with signal transduction mediated by the activation of JAK/STAT. Mechanisms that result in the transcription of genes for the production of IFN are produced subsequently.")
The 3 types of IFN with their respective intracellular signaling pathways. Type I, II y III IFNs are coupled through different receptors (IFNAR, IFNGR and IFNLR, respectively) with signal transduction mediated by the activation of JAK/STAT.
Mechanisms that result in the transcription of genes for the production of IFN are produced subsequently.
IFN I induces a variety of biological effects that can increase the autoimmunity through an alteration of the function of the key effector cells, such as B cells, T cells and DCs. For example, in vitro, IFN-α promotes the differentiation of autoreactive B cells and regulates the secretion of the “B-cell activating factor” (BAFF) and the “B cell proliferation factor” (APRIL, a proliferation-inducing ligand), allowing the activation, differentiation and proliferation of B cells.38 In some studies, the IFNs I directly induce the activation of pDCs through the expression of MHC-II, and costimulatory markers such as DC 40, DC 80, DC 86 and the production of several cytokines that stimulate the differentiation of monocytes and immature DCs into effective antigen-presenting cells.39 IFN-α also induces the differentiation of virgin T cells into helper T cells.31 In addition, IFN-α causes the inactivation of the Tregs by the regulation of intracellular AMP and the regulation of the T-cell receptor signaling, and stimulates the generation of lymph node-resident follicular T cells.40
IN SLE, the increased levels of IFN-α are intimately associated with manifestations of the disease, including the production of autoantibodies (for example: anti-ADN) and renal, hematological and central nervous system manifestations.41 In an experimental BXSB model, which produced a transient decrease of the pDCs, it led to clinical improvement, which coincided with the reduction of the genetic transcription of the IFNs, especially IFN-α.42 Similar protective effects were also observed in the transcription factor TCF4, which lacked the toll-like receptor (TLR) 7, and in lupus models B6.Sle1.Sle3 in which the pDCs were functionally inhibited by the suppression of the gene transcription factor E2-2.43
Altogether, a continuous activation of the IFN I system forced by genetic and immunological factors in SLE will lead to an autoimmune response and chronic inflammation.
The role of toll-like receptors (TLRs)Growing evidence supports the idea that the activation of TLRs plays an important role in the maintenance and progression of the disease. TLR7 and TLR9 are particularly relevant for SLE, and the stimulation through these receptors leads to very high levels of production of IFN-α. Exogenous microorganisms that act through these TLRs exacerbate the disease, and this is consistent with the association observed in outbreaks of disease established with viral infections. These circuits are, therefore, endogenous inductors of IFN-α, and the resulting production could perpetuate the autoimmune process.37,44
The most potent stimuli for the synthesis of IFN I are viral nucleic acids. Therefore, the rig-like receptors (RIG), and the TLRs 3, 7, 8 and 9 in the endosomal vesicles, recognize viral nucleic acids and initiate signal transmission pathways that activate the family of IFN transcription factors.
The myeloid differentiation factor 88 (MyD88) is a protein present in the majority of the TLR signaling pathways.45 Since both TLR7 and TLR9 use this protein, MyD88 is a target for the treatment of SLE. The inhibition of MyD88 in mice shows a marked decrease in autoimmune diseases such as, for example, lupus nephropathy.46 Teichmann et al. investigated more thoroughly the pathogenic mechanism of MyD88 in different types of cells, revealing that MyD88 regulates the B cells.47
Another potential target in the inhibition of TLR signaling in SLE are the kinases associated with IL-1R (IRAK) IRAK1 and IRAK4, which are activated by MyD88 and in turn activate the TRAF6 in TLR signaling. All TLRs, except TLR3, require IRAK and MyD88 for their signaling.48 Patients with deficiency of IRAK4 or MyD88 do not show production of autoantibodies in blood, and therefore they do not develop autoimmune diseases, suggesting that the inhibition of these pathways is essential in the pathogenesis of SLE (Fig. 2).
 use MyD88 as the signal transduction pathway, resulting in the production of IFN I and inflammatory cytokines. Some small molecule oligonucleotides or antibodies have been subject of research to inhibit these pathways.")
Potential objectives in the TLRs pathway in SLE. The TLRs (except TLR3) use MyD88 as the signal transduction pathway, resulting in the production of IFN I and inflammatory cytokines. Some small molecule oligonucleotides or antibodies have been subject of research to inhibit these pathways.
One of the objectives is the therapeutic inhibition with monoclonal antibodies focused on IFN-α. Sifalimumab, an anti-IFN-α monoclonal antibody, was evaluated, and after its administration it caused a decrease in the levels of IFN, which was more marked in the patients who were in phase I of the clinical trial with a SLEDAI score higher than 5 (moderate), than in the patients in phase II with a SLEDAI score higher than 11 (severe).49,50 It was observed an evident clinical response in the skin and joint manifestations.
Clinical efficacy was observed in patients with low serum values of IFN-α, suggesting perhaps that it can be achieved a more efficient neutralization of the activity of IFN-α in patients with significant clinical manifestations but with relatively low production of this IFN.51 It should be noted that all patients that had received single or multiple doses of rontalizumab (another type of anti-IFN-α monoclonal antibody) recovered the IFN levels 6 months after the last dose.
An interesting strategy has been reported in preclinical studies and more recently in a study in humans of the so called IFN-kinoid, a recombinant complex of IFN-α.52 The injection of IFN-kinoid resulted in a T-cell dependent production of specific IFN-α neutralizing antibodies that, in turn, decreased the expression of IFN I stimulating genes.52
In view of the suggestions that IFN-α and IFN-β might contribute to the pathogenesis of SLE, the blockade of IFNAR could provide a more effective therapy for the systemic autoimmune disease than those monoclonal antibodies that only target one subtype (Fig. 1).
The disease observed in BXSB mice with lupus that receive a specific anti-IFNAR antibody, together with data of mice from New Zealand genetically deficient in IFNAR, support this therapeutic approach.53 A monoclonal antibody specific for the subunit 1 of IFNAR, MEDI-546, prevents the association of subunit 1 of IFNAR with subunit 2, thereby blocking successive signaling processes. In contrast with the partial effects on the gene expression stimulated by IFN-α in the studies of sifalimumab and rontalizumab, this anti-IFNAR drug resulted in an almost complete inhibition of the expression of IFN stimulating genes (ISGs) in peripheral blood.54 This more complete inhibition of the IFN I pathway should be carried out with caution, taking into account the central role of IFN-α in the defense of the host during the acute phase of viral infection.
Hydroxychloroquine (HCQ), a drug used to treat malaria, is an antagonist of TLRs 7, 8, 9. The activity of HCQ has been attributed to the reduction of endosomal acidification, which is required for the activation of TLRs. More recent evidence suggests that HCQ binds directly to nucleic acids, causing structural modifications.
Numerous therapeutic agents under development direct their attention to the TLRs, or to their molecules, including oligonucleotides and small molecule inhibitors. Several oligonucleotides act as TLR antagonists The study of the drug DV-1179, a dual antagonist of TLR 7/9, was evaluated and tolerated in phase I of research, but there was no evidence of a reduction in the IFN regulatory genes.55
Preclinical studies with another dual antagonist of TLR 7/9, the IRS-954, showed inhibition of the production of IFN in pDCs in response to DNA/RNA viruses, decrease of circulating immune complexes and efficacy in murine models.56
Interestingly, an increase in the survival of pDCs mediated by TLR 7 and TLR 9 was also reversed with the treatment with IRS-954 in mice with lupus.57
Another compound, OMI-3100, demonstrated that not only inhibits IFN-α but also IL-17 from peripheral blood mononuclear cells (PBMCs).58
A TLR 7, 8 and 9 antagonist, called OMI-8400, showed efficacy in animal models, and it is currently in phase I for SLE.27,28 Both OMI-3100 and OMI-8400 have been well tolerated and, curiously, they were effective in phase II trials in psoriasis, another autoimmune disease associated with IFN.59,60
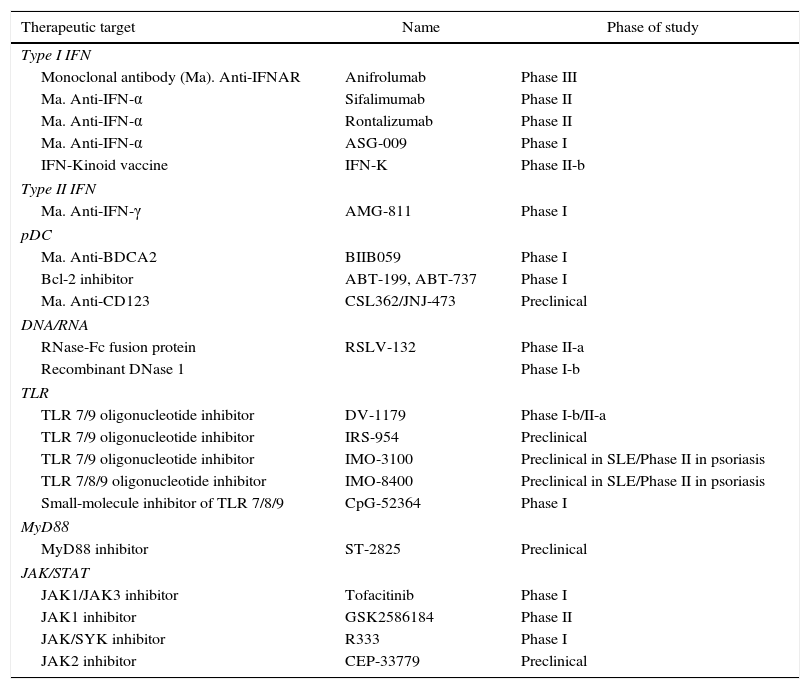
The small molecule inhibitors have the potential advantage of their oral availability, and the compounds have been designed to target TLRs and signaling proteins of this pathway, such as MyD88. The quinazoline derivative, CpG-52364, a small molecule inhibitor of TLR 7, 8 and 9, has demonstrated to be safe and more effective than HCQ in preclinical studies with animals,61,62 which have completed the phase I in clinical trials in SLE, although the results have not been yet reported.63 The inhibitor of the dimerization of MyD88, ST-2825, interferes with the activation of IRAK 4 and IRAK 1 through MyD88, and inhibits the production of proinflammatory cytokines and the proliferation and differentiation of B cells induced by TLR 964,65 (Table 1).
Therapeutic targets of the interferon pathway in SLE.
| Therapeutic target | Name | Phase of study |
|---|---|---|
| Type I IFN | ||
| Monoclonal antibody (Ma). Anti-IFNAR | Anifrolumab | Phase III |
| Ma. Anti-IFN-α | Sifalimumab | Phase II |
| Ma. Anti-IFN-α | Rontalizumab | Phase II |
| Ma. Anti-IFN-α | ASG-009 | Phase I |
| IFN-Kinoid vaccine | IFN-K | Phase II-b |
| Type II IFN | ||
| Ma. Anti-IFN-γ | AMG-811 | Phase I |
| pDC | ||
| Ma. Anti-BDCA2 | BIIB059 | Phase I |
| Bcl-2 inhibitor | ABT-199, ABT-737 | Phase I |
| Ma. Anti-CD123 | CSL362/JNJ-473 | Preclinical |
| DNA/RNA | ||
| RNase-Fc fusion protein | RSLV-132 | Phase II-a |
| Recombinant DNase 1 | Phase I-b | |
| TLR | ||
| TLR 7/9 oligonucleotide inhibitor | DV-1179 | Phase I-b/II-a |
| TLR 7/9 oligonucleotide inhibitor | IRS-954 | Preclinical |
| TLR 7/9 oligonucleotide inhibitor | IMO-3100 | Preclinical in SLE/Phase II in psoriasis |
| TLR 7/8/9 oligonucleotide inhibitor | IMO-8400 | Preclinical in SLE/Phase II in psoriasis |
| Small-molecule inhibitor of TLR 7/8/9 | CpG-52364 | Phase I |
| MyD88 | ||
| MyD88 inhibitor | ST-2825 | Preclinical |
| JAK/STAT | ||
| JAK1/JAK3 inhibitor | Tofacitinib | Phase I |
| JAK1 inhibitor | GSK2586184 | Phase II |
| JAK/SYK inhibitor | R333 | Phase I |
| JAK2 inhibitor | CEP-33779 | Preclinical |
The author declares he does not have any conflict of interest.
Please cite this article as: Encalada-García C. Células dendríticas e interferones en el lupus eritematoso sistémico. Rev Colomb Reumatol. 2017;24:177–184.
