








Infectious bursal disease virus (IBDV) is the etiological agent of a highly contagious and immunosuppressive disease in chickens. In poultry farms, the level of anti-IBDV antibodies of numerous serum samples must be monitored using fast and simple methodologies. Therefore, the aim of this study was to develop an enzyme-linked immunosorbent assay (ELISA), in an indirect format, using a version of mature viral protein 2 (VP2) of IBDV as coating agent. This recombinant fusion protein (His-VP2) was expressed at high levels in Escherichia coli. Bacterial inclusion bodies containing His-VP2 were successfully recovered using a simple, inexpensive and efficient method, a further purification of recombinant protein by affinity chromatography using immobilized metal chelates being unnecessary. After the VP2-ELISA was optimized, its performance was evaluated using preanalyzed sera from uninfected specific pathogen-free chickens and broilers vaccinated against IBDV in poultry farms, using a commercial ELISA kit. Based on these results, the developed assay proved to be sensitive, specific and in high agreement with the kit available on the market. In addition, the in-house ELISA demonstrated to be reproducible by intra-assay and inter-assay variability studies. In conclusion, VP2-ELISA could be an efficient and low-cost alternative diagnostic method to detect antibodies to IBDV in the poultry industry.
El virus de la enfermedad infecciosa de la bolsa (IBDV) es el agente etiológico de una enfermedad altamente contagiosa e inmunosupresora que afecta a pollos jóvenes. En las granjas avícolas es necesario monitorear el nivel de anticuerpos anti-IBDV de numerosas muestras de suero y para ello se requieren metodologías rápidas y sencillas. Por este motivo, el objetivo del presente estudio fue desarrollar un enzimoinmunoensayo (ELISA) en formato indirecto, utilizando como reactivo antigénico una versión recombinante de la proteína viral 2 madura (VP2) de IBDV. Esta proteína de fusión (His-VP2) se expresó en Escherichia coli en niveles elevados. Los cuerpos de inclusión bacterianos que contenían dicha proteína se purificaron utilizando un método simple, económico y eficiente y no fue necesario realizar una purificación adicional de la proteína recombinante mediante cromatografía de afinidad con iones metálicos inmovilizados. Después de establecer las condiciones óptimas del VP2-ELISA, se evaluó su desempeño utilizando sueros provenientes de pollos libres de patógenos específicos (SPF) como así también de pollos parrilleros de granjas avícolas vacunados contra IBDV y previamente analizados con un kit de ELISA comercial. Los resultados demostraron que el ensayo desarrollado es sensible, específico y tiene alta concordancia con el ELISA disponible en el mercado. Además, mediante estudios de variabilidad intra- e inter-ensayo se demostró la reproducibildad del ELISA desarrollado. En conclusión, el ensayo VP2-ELISA podría ser un método de diagnóstico alternativo, eficiente y de bajo costo para detectar anticuerpos contra IBDV en procesos productivos de la industria avícola.
During the last decade, poultry meat production in Argentina experienced a sustained growth due to the increase in local consumption and the exports of chicken (whole or in pieces) and poultry by-products. This growth was accompanied by important sanitary improvements in the producing farms; however, it is important to continue working hard on the health of the chain to ensure the quality of the products in the markets and avoid economic losses in the national poultry sector (https://www.magyp.gob.ar/sitio/areas/aves/informes/boletines/_archivos//000001Anuario%20Avicola%202022.pdf). In this context, the development of our own biotechnological tools is a priority to monitor the sanitary condition of the avian populations in productive processes.
Infectious bursal disease virus (IBDV, also known as Gumboro disease virus) is one of the main pathogens that generates significant economic losses in the poultry industry worldwide5,15,18,21,26,29. IBDV infects and destroys immature B lymphocytes within the bursa of Fabricius, the main immune organ of young birds9. The immunosuppressive status leads to an inefficient response against the vaccines used in the poultry industry as well as to the infection by opportunistic pathogens11,14,27.
The mature viral protein 2 (VP2) of 441 amino acids, is the only component of the icosahedral IBDV capsid1,3. Furthermore, VP2 has been identified as the main immunogenic protein of IBDV due to the presence of conformational epitopes in its hypervariable region (204 to 344 residues) that elicit the induction of neutralizing antibodies in the host2,10.
In Argentinian poultry farms, the control of infectious bursal disease (IBD) is performed by biosafety measures and immunization with vaccines based on inactivated or attenuated IBDV strains17. Other vaccines employed consist of immune complexes (vaccine strains with immunoglobulins against the virus) and recombinant viral vectors (such as turkey herpesvirus, fowlpox virus or canarypox virus) to express the VP2 of IBDV in vivo22.
Enzyme-linked immunosorbent assay (ELISA) is the test most commonly used to monitor the serological status of flocks, as it is economical, sensitive, specific, quantitative and allows to process a large number of samples at the same time. In particular, for the determination of anti-IBDV antibodies, there are several commercial ELISA kits, which have shown a good correlation among the antibody titers obtained with them, and the virus neutralization values and protection8,28. For this reason, the World Organization for Animal Health (OIE) recommends the use of ELISA assays to determine the levels of the immune response against IBDV in poultry farms19. Currently, most of the ELISA kits for anti-IBDV antibody detection available on the market employ the whole virus or recombinant VP2 protein as a source of antigen12,24,25,28. All of them are manufactured abroad and must be imported.
Likewise, IBDV-VP2 protein has been expressed in several heterologous systems (baculovirus, bacteria, yeast, plants) and used in the development of ELISA, which worked adequately in the detection of anti-IBDV antibodies4,6,13,23. Based on these previous results, the purpose of the present work was to develop and validate an ELISA assay that will allow, in the near future, to monitor the antibody levels in chickens during the local production processes. For this purpose, the recombinant IBDV-VP2 protein was expressed in Escherichia coli (E. coli) and subsequently the inclusion bodies (IB) harboring the recombinant protein were purified by a simple, economical and efficient method. The product obtained was used as antigen in the setup of an indirect ELISA for the detection of anti-IBDV antibodies in chicken sera. The results obtained were compared with those of a commercial ELISA kit.
Material and MethodsCloning of the VP2 sequenceFor this purpose, lyophilized Cevac®Transmune vaccine powder (Ceva Salud Animal S.R.L, Argentina), containing the IBDV Winterfield 2512 strain, was resuspended in 0.15M phosphate-buffered saline (PBS) and an aliquot was used to extract RNA with TRIzol® reagent (Invitrogen, Carlsbad, CA, USA), following the manufacturer's instructions. Then, the sequence encoding the mature VP2 of IBDV was amplified using the One Step RT-PCR kit (QIAGEN®, GmbH, Hilden, German), the previously extracted RNA and a pair of oligonucleotides specifically designed for this study (forward primer: 5’-CTCGACATGACAAACCTGCAAGATCAAACCC-3’, XhoI restriction site underlined, and reverse primer: 5’-GGATTCCTATGCTCCTGCAATCTTCAGGG-3’, EcoRI restriction site underlined and stop codon in bold letters, respectively).
The amplification product (1338bp encoding the mature VP2 protein of 441 amino acids) was first introduced into the pGEMT®-Easy vector (Promega, Madison, Wisconsin, USA) and then, subcloned into XhoI and EcoRI restriction sites of prokaryotic expression plasmid pRSET-A (Invitrogen™, Waltham, Massachusetts, USA). In the construct pRSET-VP2, the heterologous DNA was positioned downstream and in frame with a vector sequence that encodes a translation initiation site (ATG codon) followed by a polyhistidine tag (His), a phage T7 gene 10 sequence (that functions as transcript stabilizer), the XpressTM epitope and the cleavage sequence for enterokinase at theN-terminal of the fusion protein of 480 amino acids, named His-VP2. The identity of the nucleotide sequence encoding this recombinant protein was confirmed using Macrogen's sequencing service (Seoul, Korea).
Recombinant VP2 protein expression and purification of inclusion bodiesThe plasmid pRSET-VP2 was transformed into E. coli BL21(DE3) pLysS competent cells and the recombinant clones were selected with ampicillin (50μg/ml) and chloramphenicol (35μg/ml) resistance in solid Luria Bertani (LB) medium (Laboratorios Britania, Autonomous City of Buenos Aires, Argentina). Then, a time course curve was performed by induction of a bacterial culture in LB medium, at an optical density (OD600)=0.5, with 1mM Isopropyl b-D-1-thiogalactopyranoside (IPTG, Genbiotech SRL, Autonomous City of Buenos Aires, Argentina), as recommended in the pRSET vector instructions. Bacteria were grown at 37°C with shaking (200rpm) and samples of 1ml were taken at 30min intervals. The analysis of expression was carried out by 12% SDS-PAGE Coomassie Brilliant Blue staining. The identity of recombinant protein was confirmed by the Western Blot assay using an anti-VP2 rabbit polyclonal31 and a His-tag mouse monoclonal (Sigma, St. Louis, Missouri, USA) as primary antibodies. The reactive bands were revealed with alkaline phosphatase-conjugated anti-rabbit and anti-mouse secondary antibodies (ThermoFisher, Rockford, Illinois, USA) respectively, and the subsequent addition of the substrate 5-bromo-4-chloro-3-indolyl-phosphate/nitro blue tetrazolium (ThermoFisher, Rockford, Illinois, USA).
Later, the solubility of the recombinant protein was analyzed. For this purpose, the bacterial pellet from an induced culture was resuspended in 500μl of TBS buffer (50mM Tris/HCl pH 7.5; 150mM NaCl), sonicated (3 pulses of 20sec at 40kHz, with 20sec interval between each sonication, in an Ultrasonic Processor, model GE 50T) and incubated with Triton X-100 at 1% v/v final concentration during 10min at room temperature (RT). After centrifugation (9300 x g for 10min), the supernatant was separated and the pellet was resuspended in 8M Urea. The presence of the recombinant protein in both fractions was assayed by 12% SDS-PAGE.
Finally, the His-VP2 protein was produced under previously established conditions and the IB housing this protein were purified as follows: the bacteria were resuspended in lysis buffer (50mM Tris/HCl pH 7.6; 150mM NaCl; 2mM EDTA; 100μg/ml lysozyme; 1mM phenylmethylsulfonyl fluoride [PMSF]), incubated for 20min at RT and sonicated on ice (at 70kHz with 4 pulses of 20sec on and 60sec off). Triton X-100 (10% v/v final concentration) was added and the solution was vigorously vortexed and centrifuged at 12,000 x g for 10min (4°C). The pellet obtained was resuspended in buffer 1 (50mM Tris pH 8.0; 1mM EDTA; 100mM NaCl; 1mM PMSF) and centrifuged again. Then, the pellet was resuspended in buffer 1 containing 2mg/ml of sodium deoxycholate. After 10min of incubation at RT, the solution was centrifuged at 100,000 x g for 5min and the pellet was resuspended in buffer 1 containing 0.5% v/v Triton X-100. The solution was centrifuged once again (100,000 x g for 10min) and the pellet obtained was washed 3 times with distilled water. The IB were resuspended in a relation 1:10 of 6M urea and PES buffer (25mM 1,4-piperazinediethanesulfonic acid, pH 6.2; 150mM NaCl; 20mM CaCl2). Finally, 10% (v/v) glycerol was added to the IB suspension and was aliquoted and stored at -20°C until use. The amount of recombinant His-VP2 present in the IB suspension was estimated by comparison with known amounts of bovine serum albumin (BSA) in 12% SDS-PAGE stained with Coomassie Brilliant Blue.
SeraIBDV negative and positive reference chicken sera, available in our laboratory, were used to establish the optimal conditions of VP2-ELISA. Thus, the anti-IBDV positive serum was previously obtained by immunization of White Leghorn specific pathogen-free (SPF) chickens (Instituto Rosenbusch S.A., Autonomous City of Buenos Aires, Argentina) with Cevac®Transmune vaccine. Negative sera (126 samples) were obtained from White Leghorn SPF chickens, in past experiments. Addditionally, a total of 280 serum samples from vaccinated chickens in commercial farms were evaluated using the in-house ELISA.
All sera used in this work were confirmed as positive or negative against IBDV using the commercial IDEXX IBD Ab Test (IDEXX Laboratories, Inc., Westbrook, USA), following manufacturer's instructions.
To verify the analytical specificity of the VP2-ELISA, chicken sera with specific reactivity for other viruses were used. In this regard, an anti-influenza virus (H7N7) rooster polyclonal antibody, available in the laboratory; five sera from White Leghorn SPF chickens vaccinated with infectious bronchitis virus (IBV) Massachusetts strain and a commercial serum against the 4/91 isolation of IBV were evaluated.
Indirect ELISA design and standardizationTo set up the optimal concentration of antigen, flat bottom microtiter 96-well plates (Microlon®, High Binding, Greiner Bio-One, GmbH, Frickenhausen, Germany) were coated with different amounts of purified and solubilized IB containing recombinant His-VP2 protein (from 0.3 to 1.6μg/well) in 0.1M carbonate/bicarbonate coating buffer, pH 9.0. After overnight incubation at 4°C, the plates were washed four times with PBS containing 0.05% Tween 20 (PBS-T) and then blocked with 5% skim milk in PBS, supplemented with 10% adult equine serum and 1% phenol red, during 2hours at RT with shaking. Then, the plates were washed again and incubated during 1h at RT with IBDV positive and negative reference chicken sera, diluted 1:100, as primary antibodies. After four washes with PBS-T, a goat anti-chicken IgG antibody coupled to horseradish peroxidase (Bethyl Laboratories Inc, Montgomery, USA), diluted 1:10,000, was added. Following 1h incubation at RT with shaking, the wells were washed (four times) and the substrate H2O2/ABTS [2,2′-azinobis (3-ethylbenzthiazoline6-sulfonic acid) diammonium salt] in citric acid buffer, pH 4.2 was added. After 30 min of incubation in the dark, the colorimetric reaction was finished by the addition of a stop solution (2% w/v sodium fluoride) and the OD was measured at 405 nm using an automated ELISA plate reader Asys Expert Plus-Biochrom (Cambridge, England).
Next, to determine the best relationship between primary and conjugated antibodies a checkerboard titration was performed. For this purpose, the anti-IBDV antibody and the anti-chicken conjugated were diluted from 1:200 to 1:600 and 1:10 000 to 1:50 000, respectively.
The cut-off value was calculated as the mean OD of the negative sera plus three standard deviations (SD) around this mean.
To check the analytical sensitivity of the VP2-ELISA, a chicken anti-IBDV serum was diluted from 1:25 to 1:1300 and each dilution was analyzed in duplicate.
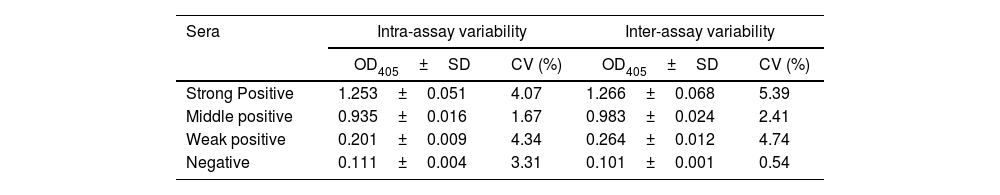
The reproducibility of the assay was verified with negative and positive (strong, moderately and weak) sera following the OIE recommendations20, with a few modifications. Briefly, the intra-assay precision (or repeatability) was determined by analyzing ten replicates of each serum samples. The inter-assay (or intermediate precision) was determined with three replicates of each serum, which were analyzed by two operators on different plates. The intra and inter-assay tests were repeated on three different days.
Data analysis and creation of graphicsThe comparison of the performance between the in-house ELISA (VP2-ELISA) and the commercial IDEXX IBD Ab Test (IDEXX-ELISA) was conducted following the OIE recommendations20. Thus, the relative sensitivity=a/ (a+c) x 100; the specificity=d / (b+d) x 100 and the accuracy=[(a+d) / (a+b+c+d)] x 100 were calculated, where a is the number of sera positive for both VP2-ELISA and IDEXX-ELISA; b is the number of sera negative for both VP2-ELISA and IDEXX-ELISA but positive for VP2-ELISA; c is the number of sera positive for both VP2-ELISA and IDEXX-ELISA but negative for VP2-ELISA and d is the number of sera negative for both assays.
Moreover, the Kappa coefficient (k) was calculated to determine the percentage of agreement between ELISA (in-house and commercial) and their capability to suitably discriminate the sera as positive or negative. K=(a+d - P) / (1 - P), where P (probability)=(a+b) x (a+c)+(c+d) x (b+d) and a is the number of sera positive for both VP2-ELISA and IDEXX-ELISA; b is the number of sera positive for IDEXX-ELISA but negative for VP2-ELISA; c is the number of sera negative for IDEXX-ELISA and positive for VP2-ELISA and d is the number of sera negative for both VP2-ELISA and IDEXX-ELISA. Kappa values between 0.21 and 0.40; 0.41 and 0.60; 0.61 and 0.80 or 0.81 and 1.00 indicate weak, moderate, good or almost perfect agreement, respectively.
The graphics were performed using the Prism version 9.5.1 computer program (GraphPad, San Diego, CA, USA).
ResultsExpression of the recombinant His-VP2 protein (His-VP2) and purification of inclusion bodiesThe sequence encoding the mature VP2 of IBDV was cloned into a pRSET-A vector, downstream of theT7 promoter and in frame with a tag of six histidines. Thus, to express the His-VP2 fusion protein, E. coli BL21(DE3)pLysS were transformed with the plasmid pRSET-VP2, induced with IPTG and the protein extracts taken at different intervals were resolved by 12% SDS-PAGE. Starting at 30min, a protein of approximately 52 kDa was evidenced by Coomassie blue staining (data not shown). This protein was recognized by antibodies with specific reactivity against VP2 and the His tag, in the Western Blot analysis (Fig. 1A and B, respectively).
 and a rabbit polyclonal serum against VP2 protein (B). The numbers above the lanes indicate the time of harvest (in min) of transformed E. coli BL21(DE3)pLysS after induction with IPTG or uninduced. SDS-PAGE solubility analysis (C) and quantification (D) of recombinant His-VP2 protein. In C, sample of insoluble (P) and soluble (S) fractions corresponding to an induced culture of transformed E. coli BL21(DE3)pLysS. In D, different amounts of purified inclusion bodies containing the recombinant His-VP2 protein were compared with known amounts of bovine serum albumin (BSA). M: protein molecular weight markers, in kDa. Arrows show His-VP2 and BSA protein bands.")
Time course curve, solubility analysis and quantification of recombinant His-VP2 protein
Western blot analysis using an anti-histidine monoclonal antibody (A) and a rabbit polyclonal serum against VP2 protein (B). The numbers above the lanes indicate the time of harvest (in min) of transformed E. coli BL21(DE3)pLysS after induction with IPTG or uninduced. SDS-PAGE solubility analysis (C) and quantification (D) of recombinant His-VP2 protein. In C, sample of insoluble (P) and soluble (S) fractions corresponding to an induced culture of transformed E. coli BL21(DE3)pLysS. In D, different amounts of purified inclusion bodies containing the recombinant His-VP2 protein were compared with known amounts of bovine serum albumin (BSA). M: protein molecular weight markers, in kDa. Arrows show His-VP2 and BSA protein bands.
Once the expression of the His-VP2 protein was confirmed, its solubility was investigated as described in Materials and Methods. The analysis conducted demonstrated the presence of the recombinant protein exclusively in the insoluble fraction (Fig. 1C).
Subsequently, the His-VP2 fusion protein was synthesized by induction of a culture for 90min The bacterial IB containing the recombinant protein were purified and then the amount of His-VP2 present in solubilized IB was estimated by comparison with known amounts of BSA in 12% SDS-PAGE (Fig. 1D). Additionally, other protein bands were not observed after IB purification demonstrating the purity of His-VP2 (Fig. 1D). This method of purification allowed to recover 32.4mg of recombinant protein (Fig. 1D) from 1 liter of bacterial culture.
Indirect VP2-ELISAThe conditions of an indirect ELISA based on purified IB containing recombinant His-VP2 protein (VP2-ELISA) were established using a checkerboard titration with different quantities of this antigen coating the wells and positive and negative sera for IBDV.
As shown in Figure 2A, the minimum amount of antigen per well that gives a good signal with the lowest cost of production was of 1.2μg. Moreover, the 1:500 dilutions of negative and positive anti-IBDV sera and the 1:10,000 dilution of the conjugated anti-chicken antibody were considered optimal to give a good signal with minimum background (Fig. 2B). Then, 126 negative sera were used to calculate the cut-off value, as described in Materials and Methods, obtaining a value of 0.1915. Subsequently, a total of 406 serum samples, classified as positive (280) and negative (126) by the IDEXX-ELISA, were analyzed with the VP2-ELISA (at a 1:500 dilution).
 and negative (■) sera for IBDV and a 1:10 000 dilution of a commercial anti-chicken antibody conjugated to peroxidase. B. Determination of suitable dilutions between primary and conjugate secondary antibodies using checkerboard titrations. Dilutions (1:200 to 1:600) of positive (curves with black symbols) and negative (curves with gray symbols) sera for IBDV were combined with dilutions (1:10 000 to 1:50 000) of the conjugate secondary antibody. In both figures the reported optical densities (OD405nm) values are the means of technical duplicates±the standard deviations. Chosen working dilutions are indicated by arrows.")
Determination of optimal concentration of VP2-ELISA reagents
A. Coating antigen titration. Different amounts of purified and solubilized inclusion bodies containing His-VP2 protein, were titrated with 1:100 dilutions of positive (●) and negative (■) sera for IBDV and a 1:10 000 dilution of a commercial anti-chicken antibody conjugated to peroxidase. B. Determination of suitable dilutions between primary and conjugate secondary antibodies using checkerboard titrations. Dilutions (1:200 to 1:600) of positive (curves with black symbols) and negative (curves with gray symbols) sera for IBDV were combined with dilutions (1:10 000 to 1:50 000) of the conjugate secondary antibody.
In both figures the reported optical densities (OD405nm) values are the means of technical duplicates±the standard deviations. Chosen working dilutions are indicated by arrows.
The mean value of OD for each serum tested is shown in Figure 3. Furthermore, these results allowed to determine a relative sensitivity, specificity and accuracy of 99%, 98%, and 99%, respectively (Table 1). Moreover, the analysis of concordance gave a k value of 0.999, which implies an almost perfect agreement between both assays.
 and positive (n=280, ♦) sera for IBDV were analyzed in duplicate and mean optical density (OD405nm) values for each serum are represented. The line indicates the cut-off value (0.1915).")
Relative sensitivity, relative specificity, accuracy and agreement of IDEXX-ELISA (Commercial IDEXX IBD Ab Test) and VP2-ELISAa
Calculations were: [(277 / 280) x 100] for relative sensitivity; [(124/ 126) x 100] for relative specificity and [{(277+124)/ (277+2+3+124)} x 100] for accuracy. The kappa coefficient value was 0.999.
In addition, the analytical specificity of the assay was also investigated using chicken sera reactive against other avian viruses. None of the analyzed sera showed reactivity against the His-VP2 as the average of their OD were lower than the cut-off value of the in-house ELISA (Supplementary Fig. S1).
In order to verify the analytical sensibility of the assay, a range of dilutions of a weak positive IBDV serum was tested. Thus, the highest dilution of this serum with an OD value above the cut-off was 1:1100 (Fig. 4).
±the standard deviation values of a weak positive serum for IBDV are graphed. The cut-off value (0.1915) is indicated with a line.")
Finally, the reproducibility of VP2-ELISA was analyzed with strong, middle and weak positive and negative sera for IBDV. As shown in Table 2, the percentage of the coefficient of variation (CV) ranged from 1.67 to 4.34% and from 0.54 to 5.39% for the intra and inter-assay, respectively. These results indicated a good reproducibility of the VP2-ELISA.
Reproducibility of VP2-ELISA.
| Sera | Intra-assay variability | Inter-assay variability | ||
|---|---|---|---|---|
| OD405±SD | CV (%) | OD405±SD | CV (%) | |
| Strong Positive | 1.253±0.051 | 4.07 | 1.266±0.068 | 5.39 |
| Middle positive | 0.935±0.016 | 1.67 | 0.983±0.024 | 2.41 |
| Weak positive | 0.201±0.009 | 4.34 | 0.264±0.012 | 4.74 |
| Negative | 0.111±0.004 | 3.31 | 0.101±0.001 | 0.54 |
Means±standard deviation (SD) of three or ten replicates are shown for intra-assay or inter-assay variability, respectively.
In the poultry farms, including Argentine chicken producing establishments, numerous serum samples must be analyzed to assess the protection level of the flocks achieved by vaccination against different pathogens; or eventually, to track an infection with field strains in non-vaccinated birds. For these purposes, ELISA tests are typically used, as they allow to analyze a considerable number of samples and to obtain results in a relatively short time.
For IBDV diagnosis, there are few commercial ELISA kits that are able to detect antibodies against the major immunogenic protein VP2. Most of these assays use IBDV purified as coating antigen with the inherent disadvantages of its production and purification. In this regard, the IBDV production requires conventional technology as well as ultra-centrifugation through a cesium chloride gradient for virus purification. In addition, careful handling and safety measures are necessary to avoid possible leaks and outbreaks of infection.
To date, there are no locally manufactured ELISA tests to determine anti-IBDV antibodies in chicken flocks; therefore, producers and diagnostic laboratories depend on the importation of these kits.
The production of the recombinant IBDV-VP2 protein (expressed in eukaryotic or prokaryotic systems) and its use as a diagnostic tool without the need to handle biohazards has been previously described in the literature. Therefore, in the present study a version of the recombinant IBDV-VP2 mature protein, N-terminally fused to a His-tag, was expressed in E. coli as IB, which were purified, solubilized and used as coating antigen to set up an ELISA assay in indirect format.
The molecular weight of the recombinant His-VP2 protein, estimated by Western Blot assay, was consistent with the size predicted from its amino acid sequence (∼ 52 kDa, Fig. 1) and was specifically recognized by both an anti-VP2 rabbit antiserum and by a commercial anti-histidine antibody (Fig. 1A and B). Additionall, the recombinant His-VP2 protein was also visible in uninduced transformed cells (Fig. 1). This fact could be due to a minimal basal production of T7 RNA polymerase prior to induction with IPTG resulting in a leak in the expression of the target protein as has been described in the literature16. Moreover, at different times post induction, faster migrating bands (∼ 28 and 30 kDa) were also revealed, which could correspond to degradation products or incomplete synthesis of the His-VP2 protein. Consequently, for antigen production, an induction period of 90min was chosen, as in this time a good concentration of recombinant protein, with the least amount of lower molecular weight bands, was observed in the expression kinetic experiments (Fig. 1A and B). After determining that the recombinant His-VP2 protein accumulates in the insoluble bacterial fraction (Fig. 1C), the purification of the IB was performed using a simple and low-cost method. When the purified recombinant antigen was analyzed and quantified by SDS-PAGE, neither contaminating protein nor bands of lower molecular weight were observed, indicating that the process was successful (Fig. 1D). These findings could be due to the fact that insoluble protein aggregates remain protected against degradation by host cell proteases during IB purification, as has been previously demonstrated7. In addition, the coated IB purified His-VP2 was specifically recognized or nonreactive in the indirect-ELISA with positive or negative chicken sera for IBDV, respectively. Moreover, a good specificity was also demonstrated through the evaluation of chicken sera reactive against some poultry viral pathogens (Supplementary Fig. S1). Taken together, these results demonstrated that processes such as fixation, through the His-tag, and subsequent elution of the recombinant protein using immobilized metal chelated chromatography could be unnecessary, thereby reducing the times and costs of purification.
In other investigations, different concentration ranges of the recombinant IBDV-VP2 protein (0 – 0.5 or 0.5 – 1.5 μg/well) were evaluated to set up the conditions of indirect ELISA assays4,30. In the present work, the optimal amount of the antigen (1.2μg/well) was defined within those reported ranges (Fig. 2A) and, therefore, the amount of antigen recovered from 1 liter of bacterial culture (32.4mg of His-VP2) was enough to coat 270 ELISA plates. Moreover,the serum dilution established for the VP2-ELISA was equal to that recommended by the IDEXX-ELISA (1:500), see Figure 2B.
Once the best conditions of the VP2-ELISA were established, the assay performance was analyzed with a panel of sera previously classified as positive and negative for IBDV by the commercial IDEXX-ELISA. In this regard, the in-house ELISA exhibited high percentages of relative sensitivity and specificity (99 and 98%, respectively, Table 1). Similar results (sensitivity and specificity of 97%) have been recently reported for an ELISA which uses as coating antigen subviral particles formed for recombinant VP2 protein expressed and purified from transgenic Nicotiana benthamiana leaves6. Other authors reported a good correlation (R2=0.837) between the IDEXX kit and an indirect ELISA which employed as antigen the precursor of the VP2 protein expressed in the baculovirus system13. Altogether, these data demonstrate the utility of the recombinant VP2 protein as a diagnostic antigen, regardless of the expression system used.
Regarding the concordance between the VP2-ELISA and the commercial IDEXX-ELISA kit, the k value showed an almost perfect agreement (Table 1), indicating that the assay developed could be used to monitor the level of immunity against IBDV in chicken populations from production farms.
The high analytical sensitivity of the in-house ELISA was determined with a weak positive serum. The OD values above the cut-off value were obtained up to a 1:1100 dilution, which is considerably higher than the working dilution (1:500), (Fig. 4).
On the other hand, the reproducibility of the VP2-ELISA was also demonstrated. In this regard, the CV values for intra (up to 4.34) and inter (up to 5.39) assays of the in-house ELISA were lower than the values (CV ≤ 10 and ≤ 20, respectively) established by the OIE20, (Table 2).
In summary, this study showed that: i) E. coli can be employed as a useful and inexpensive platform for the expression and production of the recombinant IBDV-VP2 protein; ii) the purification of bacterial IB can be conducted with a simple and low-cost method; iii) the purified and solubilized IB containing the recombinant antigen are able to adequately distinguish between positive (including weak) and negative sera for IBDV in an indirect ELISA test; iv) the developed ELISA is almost as sensitive and specific as the commercial IDEXX-ELISA.
In conclusion, the VP2-ELISA developed in the present work demonstrated to be a sensitive, specific and reproducible assay. The national manufacturing of this assay could be scaled to monitor the IBDV serostatus in poultry farms.
Ethics approvalNo ethical approval was required in this study.
Competing interestsThe authors have no relevant financial or non-financial interests to disclose.
Conflict of interestThe authors declare that they have no conflicts of interest.
The authors are very grateful to Ezequiel Nuguer for data analysis assistance, Laura Pascual from SerVetyA Poultry Laboratory, Argentina, for the supply of sera and their analysis with the commercial ELISA IDEXX Infectious Bursal Disease Virus Antibody Test Kit, Ariel Vagnozzi and Ruben Pérez for provinding the IBV-specific chicken sera, Victoria Pugliese for English language editing and Carlos Palacios for graphics support.
This work was supported by the Animal's virology studies Foundation and the National Scientific and Technical Research Council of Argentina (CONICET) (grant PU-E-22920180100014CO to ICT-MILSTEIN). These institutions provided only financial support of the research.
