




Rett syndrome (RS) is an X-linked neurodevelopmental disorder characterised by the loss of speech and functional manual skills, together with development of stereotypic movements and gait alterations. It was first described by Andreas Rett1 in 1966. In the majority of cases, it is associated with mutations that cause loss of function of the gene encoding methyl-CpG-binding protein 2 (MECP2),1 which participates in the development of motor and cognitive function. Other genes involved in the aetiopathogenesis of RS are CDKL5 and FOXG1, which participate in neurodevelopment.2
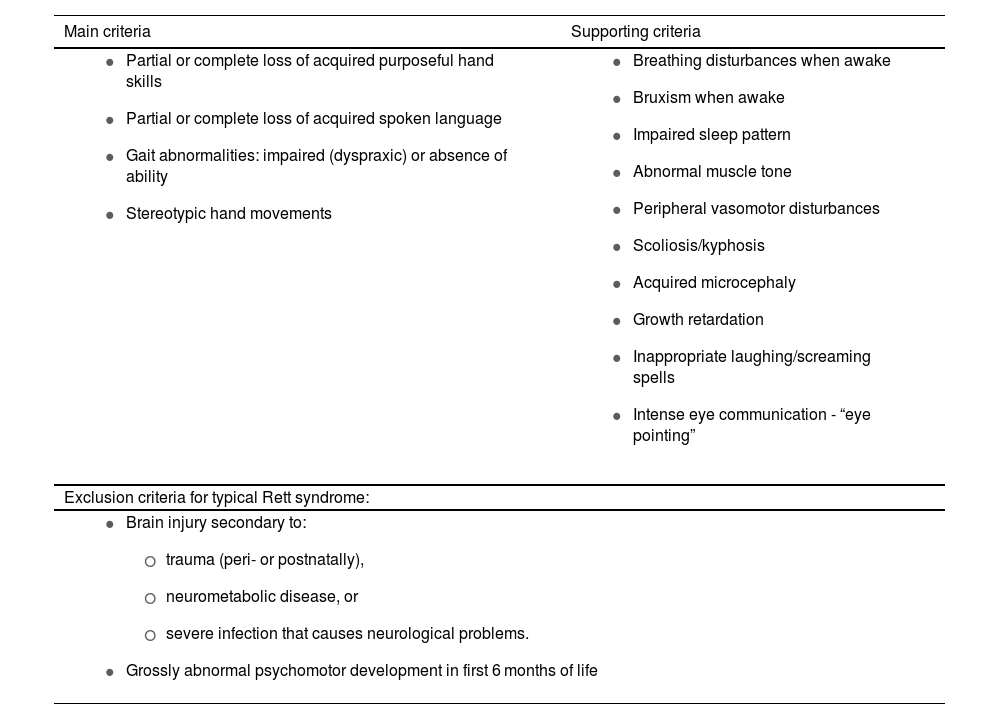
RS is one of the most frequent causes of intellectual disability in women, with an incidence of 1 case per 10 000–15 000 live female births.3 Clinical diagnosis is based on 4 major criteria and 11 supporting criteria enabling differentiation between typical and atypical forms (Tables 1A and 1B).3,4
Several clinical variants have been included in atypical RS, such as the preserved speech, late regression, congenital, and early epilepsy variants. Unlike typical RS, diagnosis of the various atypical variants may be challenging, as they are frequently diagnosed initially as autistic spectrum disorders or intellectual disability of variable severity, given the wide range of phenotypes.
We present the case of a 13-year-old girl with no relevant history who had been under follow-up by the paediatric neurology department since the age of 6 years. She was initially assessed due to procedural learning disorder. Complementary tests were performed to evaluate learning disorders. EEG, neuroimaging studies, and array comparative genomic hybridisation revealed no alterations. The patient started psycho-paedagogical therapy and subsequently showed stable progression at follow-up consultations. She developed focal seizures at the age of 8 years, and was diagnosed with epilepsy; treatment was started with an antiepileptic drug in monotherapy.
At the age of 10 years, she was admitted due to subacute signs of fluctuating level of consciousness, associated with hyperactivity and impulsiveness, and psychomotor agitation. Due to suspicion of autoimmune encephalitis, the patient started treatment with corticosteroids and gamma globulin, which were suspended after negative results were obtained in the CSF autoimmune analysis. An interictal EEG study revealed frequent epileptiform paroxysms. Suspecting non-convulsive status epilepticus, we prescribed antiepileptic treatment (valproic acid and lacosamide), together with haloperidol and clobazam, due to auto- and hetero-aggressive behaviour and severe psychomotor agitation. She was referred to the child and adolescent psychiatry department for follow-up.
The patient displayed progressive cognitive impairment, with severe attention and executive problems, slow information processing speed, and inappropriate behaviour and disinhibition. She required significant curricular adaptation and was eventually enrolled in a special educational needs centre. The patient developed symptoms of late-onset neurocognitive regression, observed with such neuropsychological assessment scales as the WISC-IV in subsequent consultations (Fig. 1).

Our examination revealed hypomimia, good eye contact, repetitive movements of the hands (simulating prominent gross tremor), loss of interest in manipulating objects, difficulties in social interaction, and repetitive behaviours. In the most recent consultations, the patient presented bruxism when awake and scoliosis of acute onset. EEG revealed severe slowing of background activity.
Considering the complex clinical picture, with cognitive regression, epilepsy, and motor, behavioural, and speech alterations, the genetic study was expanded to include clinical exome sequencing targeting genes associated with epilepsy and intellectual disability, finally detecting a de novo mutation of the MECP2 gene, with C-terminal involvement (c.1198_1226del;p.Pro400GlyfsTer7).
Neurocognitive regression in female patients is suggestive of RS, which is usually diagnosed in the first years of life. If we were to focus exclusively on clinical criteria, our patient would not meet diagnostic criteria or fit the description of the atypical forms described to date. However, neurocognitive regression, movement disorders affecting both hands with lack of interest in manipulating objects, epilepsy, and scoliosis, together with the identification of a pathogenic mutation of the MECP2 gene, make us suspect a late-onset form of RS.
Current advances in clinical genetics and molecular biology have enabled identification of previously unclassified forms, and support clinical criteria and help in predicting prognosis. Understanding the roles of various genes involved in brain maturation and development is fundamental to understanding the aetiopathogenesis of RS and identifying therapeutic targets.
Lastly, we should stress the need of families to receive an accurate diagnosis and practical, multidisciplinary help, to promote intellectual and emotional skills and to improve the quality of life of patients and their families.
FundingThe authors declare that this study received no funding of any kind.
Patient consentThe legal guardian of the patient described gave verbal informed consent for the publication of the patient's case in this article. The authors declare that they complied with the relevant protocols on the publication of patient data, and the patient's right to privacy was respected at all times.
Ethical considerationsThe authors declare that no experimental studies involving human subjects were conducted as part of the present study.
We would like to thank the paediatric neurology and clinical genetics departments of Hospital Materno Infantil de Badajoz for their help in the drafting of this article.
Diagnostic and exclusion criteria for Rett syndrome.
| Main criteria | Supporting criteria |
|---|---|
|
|
| Exclusion criteria for typical Rett syndrome: | |
| |
Supplementary material.
