






Turner's syndrome, or monosomy X, is defined as the total or partial loss of the second sex chromosome. The clinical phenotype is highly variable and includes short stature, gonadal dysgenesis, pterygium colli, cubitus valgus and low hairline. The variable expressivity of height and other physical features may be only partially related to the chromosomal formula. Currently, the delay in the diagnosis of Turner's syndrome remains a problem, as only 15–30% of patients are diagnosed during their first year of life. Understanding its complex etiology and learning more about its clinical variability and complications will allow us to advance the therapeutic and management approach of such patients. This review summarizes the clinical characteristics of, and diagnostic tests for, Turner's syndrome and the advances in the study of its underlying genetic factors.
Turner's syndrome, or monosomy X, is defined as the total or partial loss of the second sex chromosome, either X or Y.1 Clinical phenotype is highly variable and includes short stature, gonadal dysgenesis, pterygium coli, cubitus valgus and low hairline.2,3 It was first described by Ullrich in Germany in 1930.4 In 1938, Turner's described a group of seven women, ages between 15 and 23, who presented a series of physical alterations, mainly characterized by a short size and gonadal dysgenesis.5 In 1959, Ford et al. recognized the syndrome's chromosomic base, they found that patients presented 45 chromosomes with a single X chromosome.6
Turner's syndrome occurs in 1 every 2500–3000 live births and it is the only full monosomy which is compatible with life.2–4 Advances in the study of underlying genetic advances allows for a greater understanding of associated co-morbidities. Since just 30% of TS cases are diagnosed early, a proper and timely management, with a subsequent improvement in quality of life, is not accomplished in most patients. This paper reviews the main clinical characteristics, diagnostic tools and advances in the study of its underlying genetic factors.
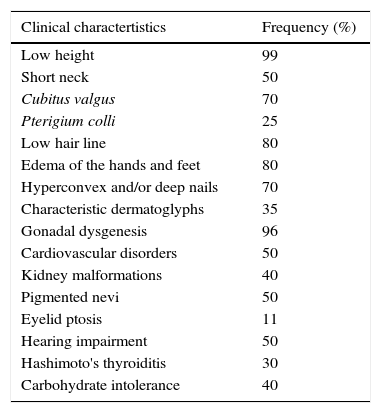
Clinical characteristicsThe variable expressivity of height and other physical features may be only partially related to the chromosomal formula, even when most recent studies do not show a clear genotype-phenotype correlation. Sometimes the phenotype may be practically normal, which occurs more frequently in cases due to partial monosomy or mosaicism.2 The frequency of structural and functional malformations are described in Chart 1.
Clinical characteristics of patients with Turner syndrome.
| Clinical charactertistics | Frequency (%) |
|---|---|
| Low height | 99 |
| Short neck | 50 |
| Cubitus valgus | 70 |
| Pterigium colli | 25 |
| Low hair line | 80 |
| Edema of the hands and feet | 80 |
| Hyperconvex and/or deep nails | 70 |
| Characteristic dermatoglyphs | 35 |
| Gonadal dysgenesis | 96 |
| Cardiovascular disorders | 50 |
| Kidney malformations | 40 |
| Pigmented nevi | 50 |
| Eyelid ptosis | 11 |
| Hearing impairment | 50 |
| Hashimoto's thyroiditis | 30 |
| Carbohydrate intolerance | 40 |
Short stature is the most constant data, it is present in over 90% of cases,2,4 it varies depending on country of origin and it is usually 20–22cm under the average population.7 TS's spontaneous growth is characterized by a moderate intrauterine growth delay, slow growth from childhood, with a progressive separation from the average population's size, absence of puberty development and bone maturation delay.
A characteristic sign is a relative widening of the thorax “shield chest”, sometimes linked to pectum excavatum. Extremity alterations are frequent, cubitus valgus and shortened metacarpal IV are both classic, more constant signs. Less frequent, however more suggesting is Madelung wrist deformity, observed from 5 to 6 years of age and characterized by a lower growth of the radius with respect to the ulna, generating a progressive dorsal deformity of the radial ulnar joint or the “Dinner fork sign”. Regarding the lower limbs, genu varum and metatarsal shortening may be found.
Some anomalies in facial bone development contribute to the configuration of the “sphynx” face. These include microrethrognatia and an underdeveloped superior maxilla, which result in an ogival palate and dental malocclusion. Other frequent facial features include palpebral ptosis, strabismus, oblique palpebral openings from top to bottom, a thin upper lip with downward commissures and a long filtrum, epicanthal folds and ear implantation with posterior rotation as a consequence of the abnormal development of the bones at the base of the cranium (Fig. 1). These bone anomalies predispose a greater risk of presenting otitis media.

One of the most typical findings during the neonatal period is lymphedema in the feet and hands, which is a result of a drainage deficit due to hypoplasia of the lymphatic vessels and is usually transitory, yet it leaves as a sequelae hypoplastic, narrow and convex nails. Said abnormal drainage has visible and persistent consequences in the neck, where pterigium colli or webbed neck, secondary to nuchal hygroma during fetal life, as well as skin folds and a low hair line and ear implantation are attributed to hygrome absorption. Pigmentary nevus are frequent, along with telangectasias and keloid scars.
Cardiovascular featuresCongenital structural heart diseases affect approximately 40% of TS patients and are an important cause of early mortality.8 These anomalies include narrowing of the aorta, a bicuspid aortic valve and abnormal pulmonary venous returns. Aortic dilatation occurs in 15–30% of girls with TS1 and depending on the size, carries a significant risk of dissection. Aortic dissection incidence in TS is estimated at 0.6–1.4% with a median age of 30–35 years of age.9
Adult patients with TS frequently show electrocardiographic abnormalities, including axis deviation to the right, T-wave abnormalities, accelerated AV conduction and prolongation of the QT-interval, often independent from structural defects.10 Essential hypertension affects up to a 25% of adolescents and 50% of adult patients.
Endocrine and reproductive featuresA greater susceptibility to endocrine and autoimmune diseases is well-documented in these patients.11 Thyroid disease has been reported in up to 30%, with hypothyroidism secondary to Hashimoto thyroiditis being the most prevalent affection. Moreover, a pro-atherogenic lipid profile and glucose intolerance with progression to type II diabetes mellitus are also frequent (2–4 times higher risk than the general population), abnormalities which, with a probable intrinsic vasculopathy, explains the high mortality rates by coronary and cerebrovascular diseases.4 Approximately 85% of patients with a chromosomal formula 45, X have a total loss of germinal cells at birth, 10–15% of these have enough germinal cells to develop a pubertal response and 5% of them have enough germinal cells to allow for pubertal development and spontaneous menstrual cycles, with a premature menopause at around 30 years of age.12–14 Natural pregnancy is produced in approximately 1–2% of all women with TS, but with a greater risk of loss.14
Other associated featuresCongenital malformations of the urinary system occur in 30–40% of patients.15 The most frequent malformations are collecting system alterations (20%), horseshoe kidney (10%) and positional abnormalities (5%). Strabismus and farsightedness occur in 25–35% of these girls, increasing the risk of amblyopia. Green-red color blindness occurs in ∼8%, a higher prevalence than that observed in women without TS due to color blindness deficiency has an X linked inheritance.16 Likewise, other monogenic disorders linked to chromosome X, like Duchenne muscular dystrophy or hemophilia (both A and B), can affect patients with TS.
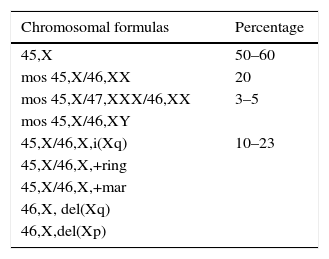
Genetic aspectsThis aneuploidy originates for the most part from a non-disjunction phenomenon which affects the paternal genome in 60–80% of cases.3 Around 50–60% of patients present a full X monosomy, which is defined as the absence of the second sexual chromosome and expressed with formula 45, X. Between 20 and 40% represent mosaics of two or more cell lines derived from the same zygote (Chart 2). Mosaicism may be limited to tissues that do not include peripheral blood, making its diagnosis difficult unless studying the affected tissue.2
Structural alterations of the X chromosome can be found in 20% of cases (Fig. 2), the isochromosome of the long arm being most frequent. Moreover, there can also be rings and X chromosome deletion of both the short and the long arms; in 3–5% we are able to find cell lines with a 46, XY complement or Y chromosome derived chromosomic markers.
 Isochromosome of the long arm of X. (B) Chromosomal mark and a ring. (C) Deletion of the short arm of the X chromosome. (D) Deletion of the long arm of X chromosome.")
The X chromosome contains 155 million pairs of bases (pb) and over 1000 genes16 while the Y chromosome has a length of 60 million pb and contains over 200 genes, from which only 48 are codifiers.17 Inactivation of the X chromosome in women allows for an equilibrium in gene doses with respect to men; however, the so-called pseudoautosomic regions (PAR1 and PAR2) escape X chromosome inactivation and their genes are counterparts to those within PAR1 and PAR2 of the Y chromosome. Hence, a proper expression and function of these genes requires both alleles. Additionally, only in said regions is there a meiotic recombination among X and Y chromosomes.
The PAR1 region covers 2.6Mb of the short arm of X and Y human chromosomes.18,19 It contains at least 24 genes, all of which escape X inactivation.6 The PAR2 region is in the distal end of the long arm, and is much smaller since it only covers 320kb7. It presents a lower recombination frequency and is composed of pseudogenes, for the most part. Four genes have been identified in this region (HSPRY3, VAMP7, IL9R and CXYorf1) located in a telomeric center direction.
The Turner's syndrome phenotype can be explained by a haploinsufficiency of the genes which are normally expressed in both sexual chromosomes and which escape X inactivation. The SHOX gene (short-stature homebox) located in Xp22.23 (PAR1) belongs to the homebox gene family, which is a transcriptional regulator and key controller of multiple processes during embryonic development. Location of the SHOX expression during embryogenesis is correlated to many phenotypic features of Turner's syndrome, since it intervenes in the development of the elbow and knee, its haploinsufficiency is associated with skeletal alterations such as cubitus valgum, genu valgum and Madelung deformity. Its expression in the first and second pharyngeal arches takes part in the formation of the upper and lower maxillary, ears and external hearing meatus,20 hence its link to a narrow palate and retrognathia. Short size is explained by the SHOX haploinsufficiency, which acts as a transcription factor over the B-type natriuretic peptide (BNP), expressed mainly in cardiac tissue and better known by its vasodilator properties. However, BNP is also expressed in other tissues, including cartilage, where it is necessary for normal chondrogenesis.21 Other genes located in the short arm of the X chromosome may contribute to the variability observed in the delay in growth. As mentioned above, BPN's predominant expression in cardiac tissue and its transcriptional control by the SHOX gene may explain cardiac alterations in these patients.
Although there are no genetic findings which explain visceral and soft tissue affectation, some phenotypical data such as lymphedema, pterigium colli and cardiac abnormalities have been linked with lymphatic hypoplasia attributable to other genes which escape X chromosome inactivation and are located in the Xp11.39 region. Immunohistochemical studies conducted in fetuses with TS showed a decrease in the expression of endothelial and vascular growth factor (FLT4, PTN63, LYVE1, Prox1) receptors, a decrease that may be linked to abnormal lymphatic genesis.22,23
Since structural alterations of the heart and vascular system are much more common in girls with cystic hygroma or lymphedema, it has been suggested that abnormal lymph nodes are involved in the development of these heart diseases. Moreover, it has been speculated that aortic coarctation is the result of abnormal lymphatic flow, which is characteristic of TS. On the other hand, there is the hypothesis that myocardial hypoplasia is a primary defect caused by lymphedema; since over 90% of fetuses with TS course with hydrops, the heart weighs under the 2.5th percentile (in relation to the weight of other organs and gestational age) while an altered heart weight was less common in those fetuses with hydrops of other etiologies.24
Zinn et al. suggested a hypothesis where a still undiscovered gene located in PAR1 causes all the neurocognitive problems of TS. They identified a region of 8.3Mb Xp22.33 whose haploinsufficiency is enough for the expression of TS neurocognitive phenotypes.25,26 The VSPA (Xp22.33) gene is clearly related to visuospatial alterations of patients with TS.27 The NLGN4X gene, proximal to PAR1 but within the 8.3Mb critical region, is another candidate to explain said neurocognitive deficit. Nevertheless, it is probable for neurocognitive manifestations characteristic of TS to be of multifactorial origin, a result of interactions between genetic and hormonal abnormalities and other unknown determinants.28
Two regions of the X chromosome contain crucial genes for the development and function of the ovary: The premature ovarian failure region 1 known as POF1, located in Xq26-28, and the premature ovarian failure region II (POF2), in Xq13.3-2.229, include known genes as well as candidate genes. In POF1, there are candidate genes HS6ST2, TDPf3, and GPC3, as well as the FMR1 gene, which is clearly linked to premature ovarian failure.30 There are genes linked to ovarian failure in POF2, such as RPS4X (a gene which codifies an isoform of the S4 ribosomal protein) and DIAPH2 (Drosophila homologue diaphone).
Other genes outside of POF regions, such as DFFRX or USP9X (Drosophila fat facets related to X) in Xp11.4, ZFX (zinc finger) in Xp22.11, UBA1 (ubiquitin-like enzyme activator-modifier) in Xp11.23 and DBX (Dead/H BOX 3, X-linked) also escape X chromosome inactivation and are related to the development and maintenance of ovarian function.14
Women with TS have a greater prevalence of autoimmune diseases such as Hashimoto thyroiditis, celiac disease and ulcerative colitis. As a matter of fact, Hashimoto thyroiditis occurs in 37% of patients with TS.31 There are at least ten genes located in the X chromosome that have possible immune regulatory functions. One of them is FOXP3, which codes a transcription factor essential for natural regulatory T-cell function (a subgroup of CD4 lymphocytes), which suppresses peripheral auto-reactive T-cells. Since mutations of this gene create a syndrome called immunodysregulation, polyendocrinopathy and enteropathy linked to the X chromosome and its haploinsufficiency would explain part of the increase in the prevalence of these diseases in TS patients. It is important to mention that the risk of autoimmune diseases, especially thyroid disease, is particularly greater in patents who present an isochromosome formation in the long arm of the X chromosome (Xq).32
DiagnosisDiagnosis is suspected in the function of clinical and hormonal findings. However, it requires a confirmation, by a conventional Karyotype (GTG bands) or other cytogenetic analysis, of the subjacent aneuploidy. The cytogenetic study ought to include a recount of at least 30 metaphases in order to be able to exclude mosaics in 10% with a confidence interval of 95%.33 In those cases where there is a great suspicion of TS and the peripheral blood caryotype is normal, studying a second tissue is indicated, usually in fibroblasts, thus ruling out a mosaicism.
Approximately 3–6% of women with TS have a 45X/46, XY mosaic and with it the risk of presenting gonadoblastoma throughout their lives is between 7% and 30%.2 Hence, ruling out the existence of sequences of the SRY gene by using molecular cytogenetic techniques such as fluorescence by “in situ” hybridization (FISH) or polymerase chain reaction techniques (PCR) is important (Fig. 3).34,35
Prenatal diagnosis
There are certain ultrasonographic findings which suggest TS. An increase in nuchal translucency is seen in TS, but it can also be observed in other chromosome disorders. The presence of septated cystic hygroma makes a TS diagnosis more likely.36 Other suggestive echo-graphic findings are aorta coarctation and/or structural heart defects, brachycephaly, malformations, polyhydramnios, oligohydramnios and growth delay. Abnormal results in triple or quadruple marker analysis in maternal serum (fetus-protein, beta chorionic gonadotropin, unconjugated estriol and inhibit A) may also suggest a TS diagnosis and justify invasive studies such as chorionic villus biopsy or amniocentesis in order to perform a Karyotype or FISH2 (Fig. 3).
Neonatal screeningSub-diagnosis and delays in TS diagnosis remain a problem.37,38 Even in countries where prenatal diagnosis is common, reports show that between 15–30% of patients are not diagnosed until after 12 years of age. It is important to stress the fact that early detection allows for proper identification and timely management of related complications. There are molecular techniques which may be applied to neonatal detection of aneuploidies of sexual chromosomes, thus defeating diagnostic delay.23,39–41 However, these have not been validated in newborn population.
TreatmentTreatment for these patients ought to be multidisciplinary, with the participation of pediatricians, endocrinologists, ophthalmologists, otorhinolaryngologists, cardiologists, geneticists, psychologists, gynecologists and reproduction biologists.
Today, growth hormone and estrogen replacement therapy results are fundamental for these patients.42 Moreover, there is reliable information stating that the combination of very low estrogen doses with growth hormones during childhood can improve the final size and provide other potential benefits linked to early estrogen replacement.43
Assisted reproduction allows for patients to get pregnant, if they chose to do so; furthermore, early TS detection allows for oocyte cryopreservation consideration and later a pregnancy in adult life.
Follow-up of these patients throughout their lives is important, with the purpose of a timely detection of concurrent diseases and prevention of possible complications. In Chart 3 there is a guide of periodic evaluations and recommended complementary studies.
Multi-disciplinary management in patients with Turner's syndrome.
| All patients |
| Cardiovascular evaluation |
| Renal ultrasound |
| Hearing evaluation by a specialist |
| Screening for scoliosis/kyphosis |
| Neuropsychological evaluation; referral to support groups |
| Evaluation of growth and pubertal development |
| Age 0–4 years |
| Evaluation for hip dislocation |
| Thyroid function tests (T4, TSH) |
| Ophthalmological examination (if age ≥1 year) |
| Age 4–10 years |
| Thyroid function (T4, TSH) and Celiac Disease (TTG Ab) tests |
| Education/psychosocial assessments |
| Dental evaluation (if age >7 years) |
| Age >10 years |
| Thyroid function (T4, TSH) and Celiac Disease (TTG Ab) tests |
| Education/psychosocial assessments |
| Dental evaluation |
| Fasting glucose or glucose tolerance curve |
| Assessment of ovarian function/estrogen replacement |
| Liver function tests, lipid profile, creatinine, BUN, urea and complete blood count |
| Bone densitometry (if age >18 years) |
Turner's syndrome is a chromosomic disorder, a result of the partial or total absence of the second sexual chromosome. A relatively frequent alteration (1:2500 newborn girls), it presents a phenotypical variability which may cause a sub-diagnosis of its cases.
It is important to know the clinical and genetic aspects which will lead to a timely detection of cases, and thus a proper management of associated co-morbidities to this genetic condition.
Conflict of interestThe authors have no conflicts of interest to declare.
We want to express our sincere appreciation to Dr. Horacio Rivera Ramírez for all his support and collaboration in this review.