






Case presentation
The patient was a 7 month-old girl product of the third pregnancy of non-consanguineous parents, whose delivery was uneventful. The patient had been initially seen by a pediatrician because of macrocephalia and psycho-motor delay; no definitive diagnosis had been made. Family history was negative. The patient was first seen at the pediatric emergency room of our institution because of a febrile respiratory illness. Physical examination, routine lab tests, and a chest x-ray confirmed the diagnosis of pneumonia; rapid influenza A test was positive. The patient was admitted to the pediatrics service where medical management was initiated. During the second day of admission, she suddenly developed vomiting, neurologic collapse, metabolic acidosis, hypotension, and cardiopulmonary arrest, and was unresponsive to resuscitation maneuvers.
A complete autopsy was performed. Weight and height were within normal limits. On external examination, both coarse face and depressed nasal bridge were identified. In situ inspection of the pelvic organs was unremarkable. The lungs showed gross changes consistent with pneumonia. Hepatosplenomegaly was present. The brain weighed 831 g (normal weight according to age/height: 691 g). The most significant gross finding on autopsy was the presence of enlarged convolutions of the frontal and parietal cerebral cortex; sulci also appeared short and scanty (Figure 1). These features were more evident on cut surface. The ventricular system, the brain stem, and the cerebellum showed no abnormalities. Except for the eyes, routine representative sections of the organs were submitted for histology. The initial histopathological study demonstrated bilateral acute pneumonia as the direct cause of death. Sections from the central nervous system (CNS), the spinal cord, and sympathetic ganglia from the paravertebral region showed that neurons presented severe morphological changes. The most striking degenerative changes consisted in neuronal enlargement with broad dendritic processes lacking slender cellular borders. The normal appearance of the cytoplasm was replaced by a large amount of granular material leading to swollen neurons with displaced nuclei (Figure 2); the intracytoplasmic material was positive for periodic acid-Schiff stain with diastase digestion and luxol fast blue. In addition, both astrogliosis in the brain cortex and spinal cord and ectopic neurons in the cerebellum were seen. Similar cytoplasmic changes were identified in interalveolar and gastrointestinal macrophages. The thymus, the spleen, the liver, and the heart were also affected. Tissue from the CNS and the lungs was submitted for electron microscopy. Ultrastructural study demonstrated numerous whorled membranous and stacked intracytoplasmic structures (Figure 3), which represent massive accumulation of glycolipids. Through histological and ultrastructural findings besides the clinical picture, the diagnosis of a lysosomal storage disease affecting the CNS and viscera consistent with infantile generalized gangliosidosis was made.
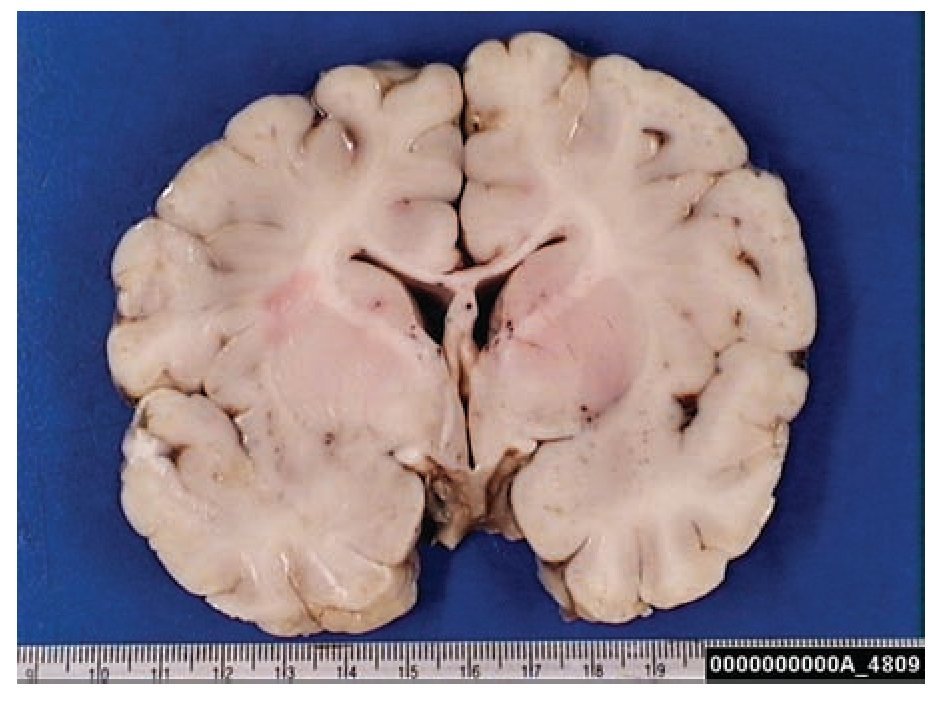
Figure 1. Gross photograph of coronal section of the brain; note the broad convolutions and short sulci.
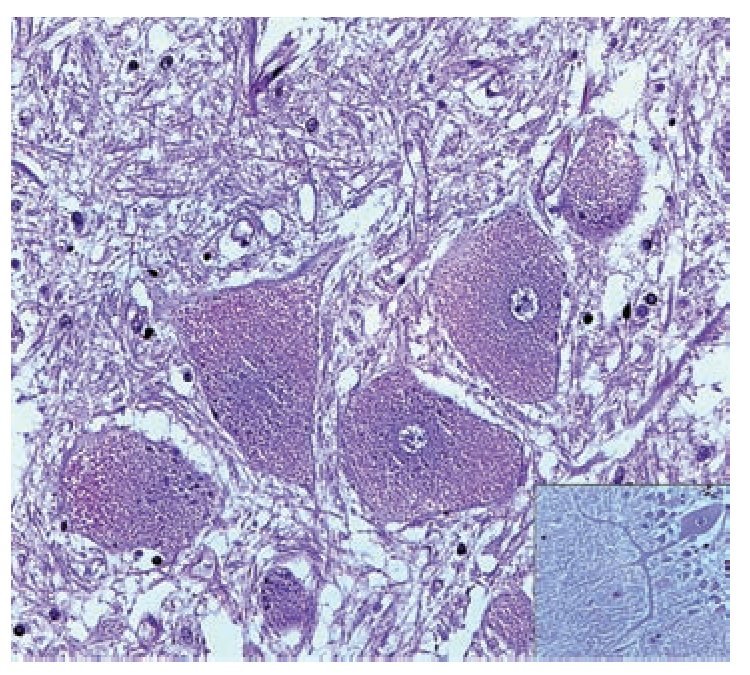
Figure 2. A group of swollen neurons are seen in this picture, gliosis (upper left) and meganeurites also are identified (inset). Hematoxilin & Eosin.
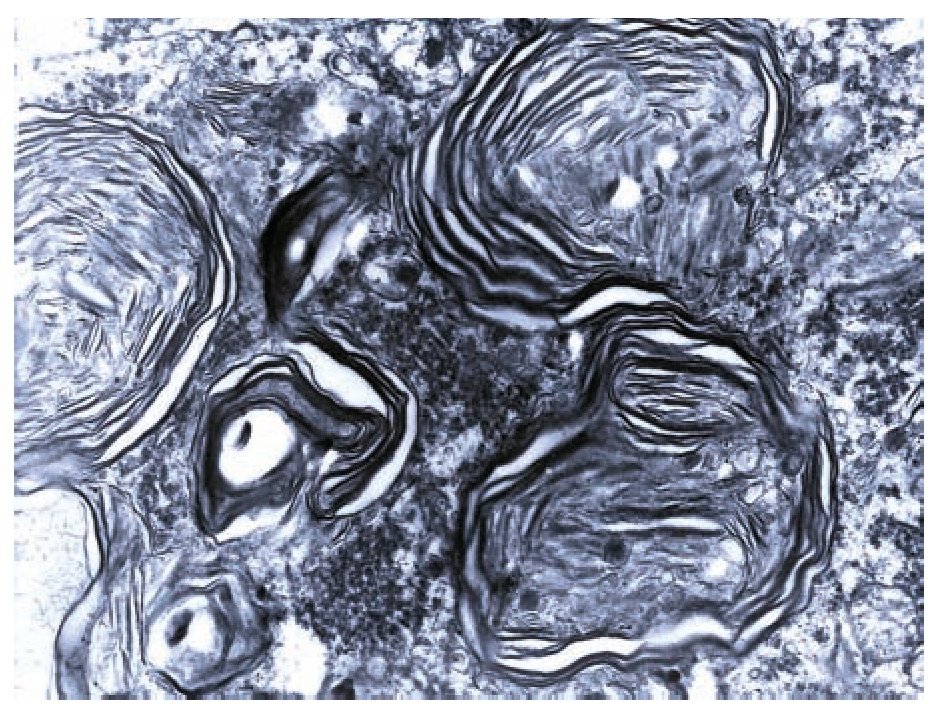
Figure 3. Numerous membranous lamellar bodies are seen in this Ultrastructure.
Discussion
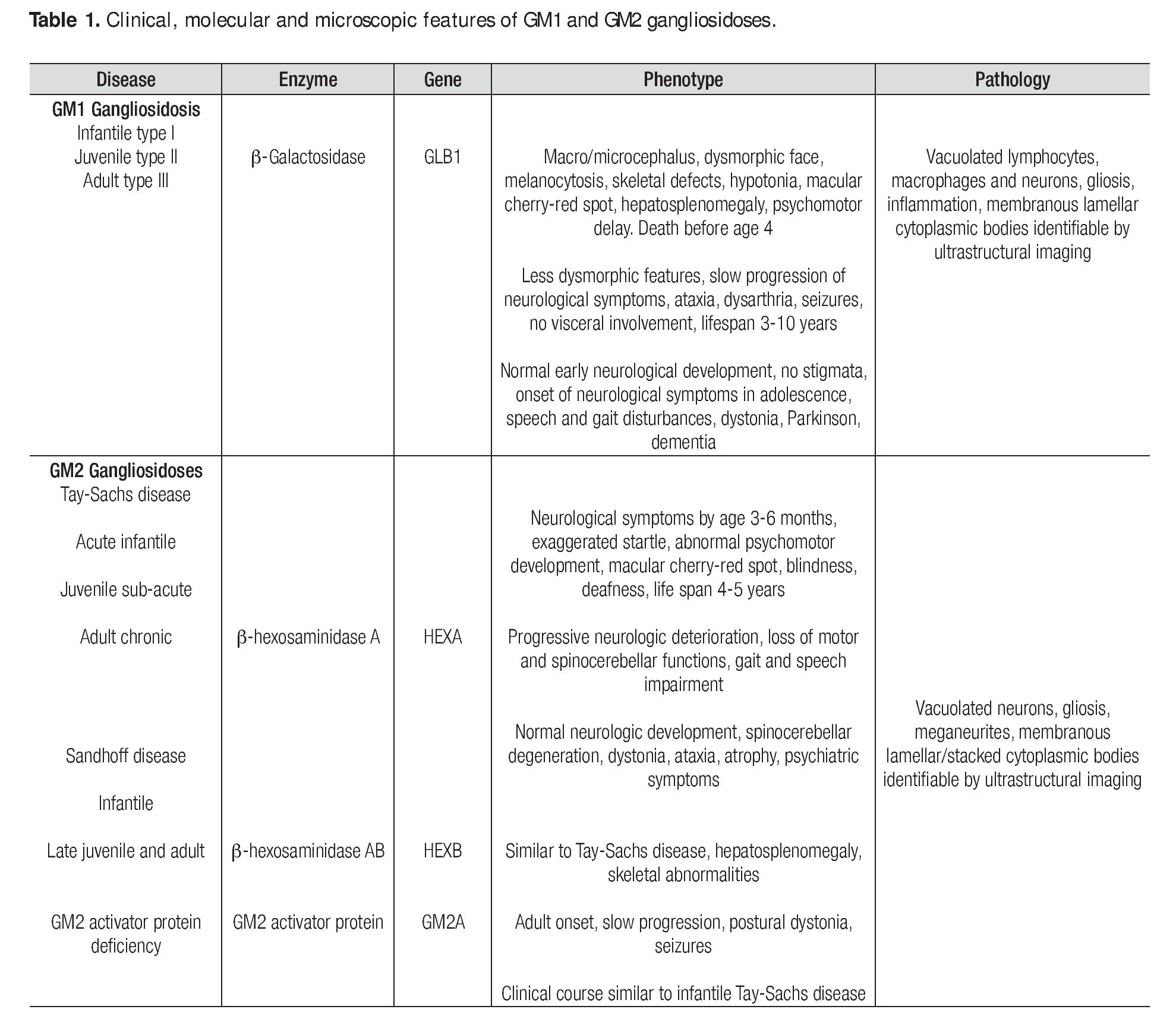
Gangliosidoses are a rare and heterogeneous group of lysosomal storage diseases inherited with an autosomal recessive trait; the metabolic disorder is caused by mutatations in any of the genes encoding enzymes involved in gangliosides metabolism. The deficiency of enzyme activity leads to accumulation of gangliosides and related glycoconjugates in the lysosomes, which causes lysosomal swelling, cellular damage, and organ dysfunction, primarily in the CNS1,2 but also in viscera.3,4 In 1942, Klenk identified a new class of carbohydrate-rich glycolipids which he named gangliosides and from which fatty acids, sphingosine, galactose, glucose, and sialic acid were isolated through hydrolysis; the gangliosides also contain hexosamine.5 Glycosphingolipids, which are essential components of every eukaryotic cell membrane, are synthesized in the endoplasmic reticulum and the Golgi apparatus and reside mainly on the external leaflet of the plasma membrane; they are internalized by endocytosis and are degraded in the lysosomes. Although once considered to be only a structural part of the plasma membrane, these molecules are now known to play important roles in a large number of regulatory events in adult life such as cell-to-cell and cell-matrix interactions. It is also thought that they are involved in many morphogenesis processes including cell signaling, growth, migration, and differentiation.6 As gangliosides are seen at the highest level of brain tissue, progressive neurological involvement is the rule. Genetic deficiency of lysosomal enzyme is varied; consequently, biochemical changes produce a significant heterogeneity in phenotypic expression. The progression of the disease depends largely on the nature of the mutation and its effect on enzyme activity. Mutations on the gene GLB1 which maps to the short arm of chromosome 3, encode the β-galactosidase enzyme and cause GM1 gangliosidosis and Morquio B disease,7,8,9,10 whereas mutations on the HEXA gene (code for a-hexosaminidase subunit-α) and the HEXB gene (code for b-hexosaminidase subunit-β) produce the GM2 gangliosidoses Tay-Sachs disease and Sandhoff disease, respectively; mutations on the GM2A gene result in GM2 activator deficiency.11,12,13 Clinical presentation, organ involvement, and age of onset show significant heterogeneity ranging from infantile-onset, rapidly progressing neurological damage, and premature death to late-onset, sub-acute, juvenile-onset, and chronic or adult forms with fewer neurological manifestations (Table 1).1,14-16 The severity of each subtype is inversely related to the residual activity of the mutant enzyme.1,11 However, the sine qua non of the disease is the progressive neurodegenerative damage that may culminate in death in the most aggressive forms of the disease.17 Gangliosi-doses are extraordinary diseases for which a panethnic background has been reported; increasing prevalence has been identified in Brazil, Japan, Malta, Cyprus, and among Jewish Ashkenazi and in Roma population.18,19,20 The incidence of GM1 and GM2 gangliosidoses has been estimated to be 1 in 100,000-200,000 live births1 and 1 in 222,000-422,000 live births,2 respectively. The diagnosis of a neurodegenerative disorder with an inherited metabolic background is not simple and relies mainly on a high index of suspicion and the clinical experience since most cases present with common pediatric problems such as recurrent vomiting, failure to thrive, seizures, developmental delay, and recurrent infections; nevertheless, several clinical features may suggest a lysosomal storage disease (Table 2).1,15,21 Lysosomal storage diseases share a common feature i.e., the biochemical disorder resulting in the accumulation within the lysosomes of normally degraded substrates. Skin and suction rectal biopsies are useful tools to elucidate the ultrastuctural nature of the accumulated substrate.22-24 A definitive diagnosis is reached through enzyme assays and molecular biology.16,25 Imaging may also positively contribute to the diagnosis.26

In the described case, clinical features of the patient were mild; however, post-mortem findings including light and electron microscopy were unequivocal. The neurons of the CNS appeared swollen with accumulation of glycolipids; a mild inflammatory process, gliosis, meganeurites, and neuronal ectopia were all observed. The histological changes seen in this case have been reported in the literature and are thought to account for the neuronal dysfunction. Despite this, the underlying mechanisms of the disease are not completely understood. Neuronal apoptosis, astrogliosis, microgliosis, demyelination, abnormal axoplasmic transport, and alteration in neuronal-oligodendroglial interactions have all been proposed as possible mechanisms of pathogenesis.27 More recently, the inflammatory response has been thought to contribute to the pathogenesis and disease progression.28,29 In animal models the accumulation of gangliosides activates microglia, which in turn recruit macrophages and T-lymphocytes initiating the inflammatory response.30 Gangliosidosis is currently an incurable disease and only support therapy is available; different therapeutic strategies have been explored including enzyme replacement, substrate deprivation, bone marrow transplantation, and gene therapy.31-33 Experiments in animal models using gene therapy and viral vectors have brought new hope for these patients.34,35 In the present case, the clinical and laboratory diagnostic approach was not completed since the patient had been admitted because of an acute respiratory disease and a lysosomal storage disease was not suspected; however, from an anatomopathological point of view, a differential diagnosis was carried out. Lysosomal storage diseases (affecting the CNS and viscera) other than gangliosidoses such as Niemann Pick disease and Gaucher's disease were ruled out since the clinical picture, the extension of the disease, and the histological and ultrastructural features were more consistent with a gangliosidoses.
Conclusion
Gangliosidoses are inborn errors of metabolism resulting from the defective activity of degradative enzymes in lysosomes and causing a neurodegenerative disorder. The diagnosis relies on clinical suspicion, imaging, light and electron microscopy, and enzyme assays or molecular biology. Reaching a correct diagnosis is of utmost importance for appropriate therapy and genetic counseling.
Corresponding author:
Dr. Marco Antonio Ponce Camacho.
Servicio de Anatomía Patológica y Citopatología, Facultad de Medicina y Hospital Universitario Dr. José Eleuterio González, de la Universidad Autónoma de Nuevo León. Av. Francisco I. Madero y Av. Gonzalitos s/n, Colonia Mitras Centro, C. P. 64460, Monterrey, Nuevo León, México.
Phone: 52 (01-81) 8333 8181.
E-mail:drmarcponce@hotmail.com
Recibido: enero, 2010.
Aceptado: febrero, 2010.