





Acromegaly and gigantism are due to excess GH secretion, usually by a pituitary adenoma. It is an uncommon disease.
Diagnosis is made by showing elevated GH and IGF-I levels in patients with a clinical picture suggesting the condition. Once excess GH is confirmed by biochemical tests, MRI of the hypothalamic–pituitary area should be performed to ascertain the source of excess GH.
Transsphenoidal surgery of the pituitary adenoma is the treatment of choice. However, introduction of new drugs has changed the treatment sequence in recent years. Medical treatment with somatostatin analogs may be indicated as primary treatment in patients in whom surgery is not expected to be curative or is contraindicated. The GH receptor antagonist should be used in patients not controlled after surgery who do not adequately respond to somatostatin analogs.
Radiotherapy would be indicated in patients not controlled after surgery and medical treatment or with large tumor remnants after surgery.
La acromegalia y el gigantismo se deben a la producción excesiva de GH, generalmente por un adenoma hipofisario. Es una enfermedad poco frecuente.
El diagnóstico se realiza ante un paciente con un cuadro clínico sugerente con la demostración de concentraciones de GH e IGF-I elevadas. Tras la confirmación bioquímica del exceso de GH debe realizarse una RM del área hipotálamo-hipofisaria a fin de confirmar el origen del exceso de GH.
El tratamiento de elección es el quirúrgico del adenoma hipofisario mediante cirugía transesfenoidal, si bien en los últimos años los avances en cuanto a la aparición de nuevos fármacos han modificado la secuencia terapéutica. El tratamiento médico con análogos de somatostatina puede estar indicado como procedimiento primario en pacientes no subsidiarios de curación tras cirugía, o en aquellos casos en que esta esté contraindicada. El antagonista del receptor de GH debe utilizarse en pacientes no controlados tras cirugía que no responden de forma adecuada a análogos de somatostatina.
La radioterapia estaría indicada en aquellos casos no controlados tras tratamiento quirúrgico y médico o en aquellos pacientes con grandes restos tumorales tras el tratamiento quirúrgico.
Acromegaly and gigantism are due to excess GH secretion, usually by a pituitary adenoma. Diagnosis is invariably preceded by approximately 10 years of unknown disease.1
Pituitary adenoma accounts for approximately 15% of intracranial tumors. The incidence of acromegaly is approximately five cases per million people, per year, while the prevalence rate is 60 cases per million people.
Somatotroph adenomas are of monoclonal origin and develop from genetic changes. Hypothalamic and paracrine GHRH and somatostatin, like growth factors, promote the expansion of tumoral somatotroph cells.1
More than 90% of patients with acromegaly have a monoclonal benign pituitary adenoma surrounded by non-hyperplastic pituitary tissue. Densely granulated adenomas grow slowly and occur in patients older than 50 years. Poorly granulated adenomas grow more rapidly and occur in younger patients. Approximately 25% of GH-secreting adenomas co-secrete prolactin. These include dimorphic adenomas with GH and prolactin cells, mammosomatotroph monomorphic adenomas, and adenomas of more primitive acidophilic cells. Multicellular or unicellular mixed immunoreactivity is common, especially for the alpha subunit of glycoprotein hormones. The secretion of other clinically relevant hormones rarely occurs.
Pituitary adenomas occurring in children before growth is completed cause gigantism. Pituitary gigantism is very rare. In a large series of 2367 children and adolescents with pituitary adenomas, only 0.6% had gigantism.2
More than 70% of somatotroph adenomas are macroadenomas at diagnosis, but somatotroph cell carcinoma is exceptional. Ectopic GH secretion is also exceptional. Familial acromegaly syndromes are very rare. Excess GHRH production of hypothalamic (usually gangliocytomas) or peripheral central origin may lead to somatotroph cell hyperplasia and acromegaly.
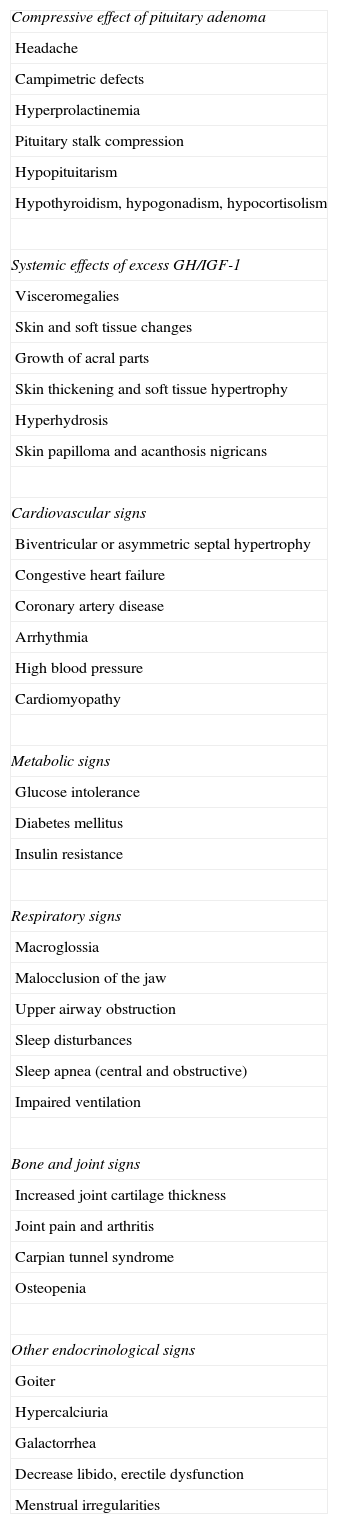
Clinical signsThe clinical signs of acromegaly include facial changes, the growth of acral parts, prognathism, hyperhydrosis, headache, paresthesia, sexual dysfunction, high blood pressure, goiter, soft tissue growth, joint pain, symptoms of hyperglycemia, bone and joint disease, cardiomyopathy, heart failure, sleep apnea, and respiratory failure. Symptoms derived from local manifestations of the tumor, such as visual disturbances, also occur. Visceromegalies frequently occur as goiter, hepatomegaly, splenomegaly, and macroglossia. Subtle signs of growth of acral parts, bone growth, and soft tissue enlargement inexorably develop over the years. Bulging of the forehead, prognathism, skin thickening, and increased size of shoes and rings (Grade C) are particularly characteristic (Table 1).
Clinical and laboratory signs of acromegaly.
| Compressive effect of pituitary adenoma |
| Headache |
| Campimetric defects |
| Hyperprolactinemia |
| Pituitary stalk compression |
| Hypopituitarism |
| Hypothyroidism, hypogonadism, hypocortisolism |
| Systemic effects of excess GH/IGF-1 |
| Visceromegalies |
| Skin and soft tissue changes |
| Growth of acral parts |
| Skin thickening and soft tissue hypertrophy |
| Hyperhydrosis |
| Skin papilloma and acanthosis nigricans |
| Cardiovascular signs |
| Biventricular or asymmetric septal hypertrophy |
| Congestive heart failure |
| Coronary artery disease |
| Arrhythmia |
| High blood pressure |
| Cardiomyopathy |
| Metabolic signs |
| Glucose intolerance |
| Diabetes mellitus |
| Insulin resistance |
| Respiratory signs |
| Macroglossia |
| Malocclusion of the jaw |
| Upper airway obstruction |
| Sleep disturbances |
| Sleep apnea (central and obstructive) |
| Impaired ventilation |
| Bone and joint signs |
| Increased joint cartilage thickness |
| Joint pain and arthritis |
| Carpian tunnel syndrome |
| Osteopenia |
| Other endocrinological signs |
| Goiter |
| Hypercalciuria |
| Galactorrhea |
| Decrease libido, erectile dysfunction |
| Menstrual irregularities |
Acromegaly has an insidious onset, which results in an 8- to 10-year delay in diagnosis. The clinical signs in each patient depend on GH and IGF-1 levels, age, tumor size, and delay in diagnosis.3,4
DiagnosisMost patients have clear clinical signs.
A diagnosis of acromegaly requires the demonstration of high GH and IGF-1 levels. GH levels are tonically elevated in acromegaly, and a random GH level less than 0.04μg/L therefore rules out diagnosis, but a high random level does not imply the presence of acromegaly.
Biochemical diagnosis is made by measuring fasting IGF-1 levels and GH levels before and after a 75g oral glucose tolerance test (OGTT). Diagnosis is sometimes complicated by physiological GH changes and by the lack of standardization of tests for measuring GH. New immunoassays based on monoclonal antibodies are more sensitive, but have significant reproducibility problems.5
After OGTT, nadir GH levels less than 1μg/L rule out acromegaly with most methods. However, if ultrasensitive methods are used to measure GH, suppression to less than 1μg/L may occur in some patients with acromegaly; with some of these methods, the suppression criterion is a circulating level of 0.4μg/L or less (depending on the test used) according to the most recent consensus on the cure of acromegaly, or even 0.3μg/L according to other authors.1,6,7 GH may not be suppressed in the presence of liver or kidney disease, poorly controlled diabetes mellitus, malnutrition, anorexia, pregnancy, or estrogen therapy, or in late adolescence (Fig. 1).

Diagnostic algorithm for acromegaly. OGTT: oral glucose tolerance test.
Thus, IGF-1 acts as a biomarker of activity of acromegaly in different situations. IGF-1 levels are relatively stable and correlate to clinical acromegaly data and to increased plasma GH levels. No additional increase occurs in plasma IGF-1 levels when GH levels are higher than 20μg/L, and subtle GH increases do not always increase circulating IGF-1 levels. Accurate IGF-1 assessment requires age-adjusted controls, because IGF-1 levels decrease 14% by decade. The measurement of circulating GH and IGF-1 levels is a supplemental tool for disease monitoring.
Pituitary magnetic resonance imaging (MRI) with gadolinium contrast is the best imaging procedure for locating the source of excess GH. This technique allows for the visualization and localization of adenomas greater than 2mm in diameter in relation to the surrounding structures. At diagnosis, more than 75% of patients have a macroadenoma (greater than 10mm in size) which grows into the cavernous sinus or suprasellar region. In those rare cases where a non-pituitary origin is suspected, computed tomography or MRI of the chest and abdomen or octreoscan scintigraphy should be performed.
TreatmentTreatment objectives in acromegaly include:
- 1.
Tumor growth control.
- 2.
Normalization of high IGF-1 and GH levels.
- 3.
Symptom control, quality of life improvement, and control of comorbidities.
- 4.
Prevention of early mortality (Grade B).
Three treatment modalities currently exist: surgery, medical treatment, and radiotherapy6,8,9 (Fig. 2).
 Of choice if there are no contraindications or intolerance. Primary treatment in those not undergoing surgery for any reason. (2) To be considered in: very elderly patients, slight GH and IGF-1 elevations, mixed tumors secreting GH and prolactin. (3) Not approved as first drug option in label. To be considered in patients with very severe clinical signs or highly increased IGF-1 levels.")
Treatment algorithm for acromegaly.
(1) Of choice if there are no contraindications or intolerance. Primary treatment in those not undergoing surgery for any reason.
(2) To be considered in: very elderly patients, slight GH and IGF-1 elevations, mixed tumors secreting GH and prolactin.
(3) Not approved as first drug option in label. To be considered in patients with very severe clinical signs or highly increased IGF-1 levels.
Surgery continues to be the treatment of choice for acromegaly in most patients (Grade B), although advances in medical treatment in recent years have changed the treatment sequence.6,8,9
Radiotherapy is currently the last step in the treatment scheme, being reserved for patients not controlled after initial medical or surgical treatment and for uncontrolled, invasive macroadenomas (Grade C).6,9
It is important that patients are cared for by a multidisciplinary team consisting of experienced endocrinologists, neurosurgeons, and radiation therapists able to recommend the most adequate treatment in each case (Grade C).6,8
SurgerySurgery is the first-line treatment in most patients. It is the procedure of choice in microadenomas, macroadenomas with compressive symptoms, and macroadenomas amenable to surgical cure (Grade A). Surgery may also be the treatment of choice for macroadenomas not amenable to cure in order to decrease tumor size and facilitate the response to supplemental treatment (Grade B).
In a recently published survey on the management of acromegaly, surgery was the treatment of choice in most patients. It was the treatment chosen by the surveyed physicians in Europe and the USA for 90% and 94% of microadenomas and 92% and 94% of macroadenomas with visual involvement respectively. This proportion decreased in macroadenomas not compressing adjacent structures and not amenable to cure after surgery.10
Advantages of surgeryTumor size decrease or total resection may induce a cure, causing a rapid decrease in hormone levels that stops disease progression and improves comorbid conditions.
In invasive macroadenomas not amenable to cure, tumor mass resection causes the rapid decompression of adjacent structures and normal pituitary tissue, preserving pituitary function and facilitating the response to subsequent medical and radiotherapeutic treatment.
Surgery allows for the collecting of tumor tissue for immunohistochemical, ultrastructural, and molecular study, which helps in selecting the best subsequent medical treatment.
Patient preparation before surgeryA detailed clinical history, including basic laboratory tests and pituitary hormone assessment, especially in macroadenomas, should be taken to confirm the need for hormone replacement of the adrenal or thyroid axis before surgery (Grade C). Cardiovascular risk and the presence of comorbidities that should be treated before surgery should also be assessed.9
Pre-surgical treatment with somatostatin analogsSome studies show that patients treated with somatostatin analogs (SSAs) before surgery achieve greater remission rates as compared to untreated patients (Grade C). However, other studies do not support these findings. In a Norwegian multicenter study, pre-treatment with octreotide for six months achieved a cure (defined as normal IGF-1 levels) in 50% of patients with macroadenoma, as compared to 16% of untreated patients. However, when GH levels after OGTT less than 1μg/L were added as a cure criterion, the difference between the two groups was not significant.11 In an additional study of 98 patients with macroadenoma treated with lanreotide for four months, remission rates after surgery were 49% and 18% in the treated and untreated groups respectively. The difference between the two groups remained significant when GH suppression after OGTT to below 1μg/L was added.12
In some studies, prior treatment with SSAs decreased the incidence of complications or hospital stay, although other studies do not support these findings.
SSAs improve cardiac function by decreasing ventricular hypertrophy and the incidence of arrhythmia, which, combined with their beneficial effects on respiratory tract tissue edema in facilitating intubation, make them a convenient treatment option before surgery (Grade C).13–15
In the abovementioned survey, 75% of participants used SSAs before surgery, 35% to decrease the risks of anesthesia, 24% to achieve prior biochemical control, and 10% based on patient preference. Although medical treatment may be indicated, additional evidence is required before it is routinely recommended for all patients with acromegaly undergoing surgery.10
Successful surgery depends on the experience of the neurosurgical team16 (Grade A). The extent of control correlates to neurosurgical experience. A neurosurgeon should perform at least 50 annual surgical procedures before being considered an expert.
Surgical procedureTranssphenoidal surgery is the surgical procedure of choice. Craniotomy is rarely indicated today in patients with acromegaly.
The new endoscopic surgical procedures, neuronavigation, the intraoperative use of MRI, or the measurement of intraoperative GH levels may improve postoperative follow-up and patient satisfaction and decrease the complication rate. Early results are encouraging but limited, and additional studies to support them are therefore required. These are expensive procedures prolonging surgery duration. Their use depends on the decision and experience of the neurosurgical team.17
ResultsThe efficacy of surgery is inversely correlated to:
- 1.
Tumor size.
- 2.
Pre-surgical GH and IGF-1 levels (Grade C).
In microadenomas, surgery achieves cure rates ranging from 75% to 95% (Grade B). Cure rates in noninvasive macroadenomas are smaller, and a normalization of IGF-1 levels is achieved in 40–68% of patients.10,11 The influence of tumor size on surgical success is controversial, but it is usually recognized that tumors greater than 2cm in size are associated with lower cure rates (Grade B). Cure rates in invasive macroadenomas are low. On the other hand, GH levels higher than 30μg/L are associated with cavernous sinus invasion and low surgical cure rates (20–50%)18–20 (Grade B).
Data from the Spanish Acromegaly Register show that 81.2% of patients were treated with surgery. The cure rate was 40.3% (defined as GH levels below 2μg/L after OGTT and/or normal IGF-1).21
Repeat surgery may be considered in patients with tumor remnants visible in MRI in whom cure is not achieved after initial surgery.3
Perioperative managementUrine output and electrolyte levels should be monitored after surgery because of the potential occurrence of diabetes insipidus, which may occur in 10–20% of patients. This is most often transient, but may be permanent in 2–7% of cases and require long-term treatment with desmopressin.22
Hyponatremia related to the syndrome of inappropriate antidiuretic hormone secretion occurs 5–14 days after surgery in 5–10% of patients. The syndrome is most often mild and only requires water restriction, but is occasionally more severe and may require the intravenous administration of hypertonic saline.22
Adrenal function should be monitored in the early postoperative period and glucocorticoid replacement therapy should be started if necessary (Grade C).
Postoperative monitoringAn early decrease in GH at 24–48h (below 2μg/L) is associated with long-term remission, but the measurement of IGF-1 is recommended between weeks 9 and 12 after surgery. Normalization of IGF-1 is associated with surgical remission. If elevated IGF-1 levels persist at 12 weeks, they should be measured again after another 12 weeks to see if late remission has occurred before starting supplemental treatment.
An OGTT with 75g for GH suppression (with measurements every 30min) should be performed from three months. GH suppression below 1μg/L is associated with a high probability of surgical remission (Grade C). GH suppression below 0.4μg/L increases test sensitivity.6,20
Postoperative MRI should also be performed from the third month after surgery to assess whether residual tumor exists.
Pituitary function tests should be done between weeks 6 and 12 after surgery to assess the need for hormone replacement therapy (Grade C).
Histological studyThe neurosurgeon should always take a sample for histological study. The proliferation index Ki67 may indicate greater or lesser aggressiveness and guide subsequent treatment. Tumors with positive immunohistochemistry for GH without clinical signs of excess GH are called clinically silent, but may have an aggressive behavior and be amenable to treatment with SSAs.
Tumors positive for prolactin may respond better to dopamine agonists. Poorly granulated tumors respond to treatment with SSAs worse than densely granulated tumors. Finally, molecular study of the different types of somatostatin receptors may serve as a guide to more effective subsequent treatment.9
Medical treatmentThe medical treatment of acromegaly may be prescribed as follows:
- 1.
As primary treatment for patients who have a significant surgical risk, in those with a low chance of cure because the tumor has extrasellar extension, without chiasm compression, and in patients who refuse surgery and choose medical treatment (Grade C).
- 2.
As supplemental treatment after surgery failure or in the interim period until radiotherapy becomes effective (Grade B).
- 3.
It may also be prescribed as pre-treatment before surgery to improve the anesthetic conditions in the patients or the results of surgery itself, or in patients in whom surgery is delayed.
There are currently three drug classes that may potentially be used: SSAs, dopamine agonists, and peripheral GH antagonists (Grade A).
Somatostatin analogsMechanism of actionSSAs, like native somatostatin itself, have an inhibitory action on GH secretion. The currently available SSAs, octreotide and lanreotide, mainly act through subtype 2 receptors and to a lesser extent on subtype 5 receptors. The response may therefore depend on the greater or lesser presence of these receptor subtypes in the tumor (Grade C).23 A new SSA under development, SOM-230 or pasireotide, shows a greater potency of action through subtype 5 receptors and a wider spectrum on subtypes 2, 3, and 1, but a poorer behavior on carbohydrate metabolism.24,25 The action of SSAs is mainly mediated through subunit Gα, inhibiting adenyl cyclase and decreasing cAMP generation. They also regulate tyrosine phosphatase activity and calcium and potassium channels.
How supplied and dosage schemesSoluble octreotide (Sandostatin®) was initially used at doses of 100μg every 6 or 8h, up to a maximum total dose of 1500μg/day. The advantages of soluble octreotide include faster action, subcutaneous self-administration, and lower costs, but patients prefer the more recent long half-life formulations, which require intramuscular or deep subcutaneous administration (lanreotide 30mg every 10–14 days), or even formulations such as octreotide LAR microspheres for suspension (Sandostatin® 10, 20, and 30mg) and lanreotide autogel (Somatulina autogel® 60, 90, and 120mg), whose mean administration frequency is every 28 days. The dosage should be individualized based on treatment response. In long-term treatments, doses may be given with time intervals up to 42 or 56 days if an effective response is seen.
The octreotide label recommends that, before treatment is started with the LAR formulation, the soluble subcutaneous form, 100μg/8h, should be used for two weeks to assess tolerability, although this may also be established with one or two doses (Grade C). The value of an acute test for predicting the future response has been questioned26,27 (Grade C). Once tolerability is shown, depot forms may be used at an intermediate dose (20mg for octreotide LAR, 0mg for lanreotide autogel) which is subsequently adjusted in accordance with the response.
During treatment with SSAs, IGF-1 or both GH and IGF-1 levels should be measured. An OGTT for GH suppression and serial GH measurements are not required (Grade D). It should be noted that IGF-1 levels reach a maximum with very high GH levels, and a delay in IGF-1 reduction should therefore be expected as GH levels decrease.
Clinical effectsSSAs have been shown to be effective for controlling the plasma levels of both GH and IGF-1 (Grade B). Soluble octreotide achieves GH and IGF-1 reduction in 50–70% of patients, with a normalization of IGF-1 in 30% of patients in whom surgery has failed (Grade B).28
As the primary treatment, SSAs allow for biochemical control (IGF-1 normal for age and sex) in up to 70% of patients and are well tolerated. A greater response to SSAs has also been seen when they are combined with debulking surgery, which may be considered for patients with advanced disease (Grade C).
As adjuvant treatment, long-acting SSAs achieve a normalization of IGF-1 in 67% of patients and normal GH levels (less than 2.5μg/L or below 1μg/L after OGTT) in 57% of patients, according to a meta-analysis of 44 trials, with a greater response at six months of treatment (Grade B).29 Octreotide LAR and lanreotide autogel have a similar efficacy profile (Grade B).
Several predictors of response have been reported. The best predictors are age and sex (better responses are seen in elderly patients and women of childbearing age or on estrogen therapy), but mainly tumor size (with better responses expected in smaller and less invasive tumors), as well as lower GH and IGF-1 levels before treatment. Other predictors of response reported include the abovementioned GH decrease following a dose of soluble octreotide (acute test), prior surgery, the uptake of labeled octreotide (Octreoscan) (Grade D), prior radiotherapy, the presence of the gsp oncogene (mutation at the Gsα complex subunit) (Grade D), a hypodense image signal in T2-weighted MRI (Grade D), as well as histopathological characteristics, because densely granulated tumors respond better (Grade C).30–35
In addition to their antisecretory effects, SSAs have been shown to be effective in reducing tumor size because of their antiproliferative action (Grade B). A meta-analysis of 14 studies concluded that SSAs achieve a significant reduction in up to almost 40% of patients, with no significant difference between fast-acting and long-acting analogs or microadenomas and macroadenomas (Grade C).36,37
However, up to one-third of patients show resistance to SSA action (Grade B). This resistance is seen as an inadequate biochemical response and a failure to achieve a tumor reduction greater than 20%, although partial responses may occur (Grade C). Resistance often depends on treatment time and dose, which may be increased up to octreotide LAR 60mg (Grade C).28,38
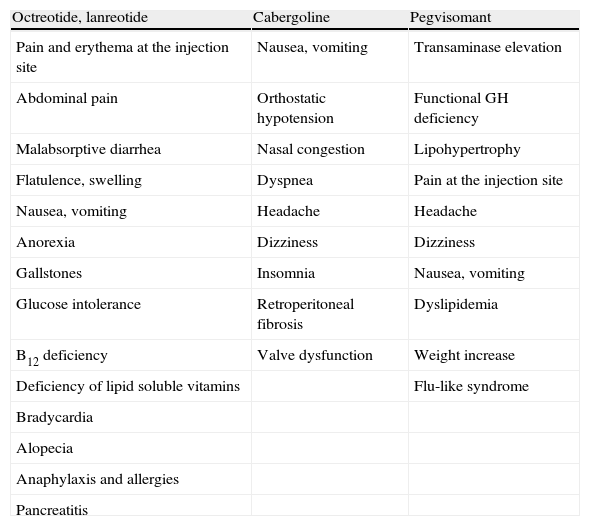
Adverse effects (Table 2)SSAs are usually well tolerated. The most common adverse effects are gastrointestinal in nature (Grade A) and include malabsorptive diarrhea, cramp-like abdominal pain, flatulence, nausea and, less commonly, constipation (Table 2).
Side effects of drugs used to treat acromegaly.
| Octreotide, lanreotide | Cabergoline | Pegvisomant |
| Pain and erythema at the injection site | Nausea, vomiting | Transaminase elevation |
| Abdominal pain | Orthostatic hypotension | Functional GH deficiency |
| Malabsorptive diarrhea | Nasal congestion | Lipohypertrophy |
| Flatulence, swelling | Dyspnea | Pain at the injection site |
| Nausea, vomiting | Headache | Headache |
| Anorexia | Dizziness | Dizziness |
| Gallstones | Insomnia | Nausea, vomiting |
| Glucose intolerance | Retroperitoneal fibrosis | Dyslipidemia |
| B12 deficiency | Valve dysfunction | Weight increase |
| Deficiency of lipid soluble vitamins | Flu-like syndrome | |
| Bradycardia | ||
| Alopecia | ||
| Anaphylaxis and allergies | ||
| Pancreatitis |
After long-term use, gallstones occur in 5–20% of patients (Grade B). Stones are usually asymptomatic, and ultrasound monitoring, with a frequency which has not been established yet, may therefore be recommended. Symptomatic gallstones should be treated with surgery or drugs that dissolve bile salts.29,39,40
Malabsorption may impair fat absorption in some patients, and decrease vitamin B12 absorption.
Hair loss and bradycardia may less commonly occur, mainly in patients who are already taking drugs inducing bradycardia (beta-blockers or calcium channel blockers).41
SSAs impair glucose tolerance and may cause diabetes mellitus or worsen already existing diabetes. In these cases, the dosage may be decreased if allowed by disease control, the GH antagonist may be substituted, or glucose control with hypoglycemic drugs may be optimized.42,43
SSAs also decrease cyclosporin absorption and delay cimetidine absorption, increase bromocriptine availability, and may interact with other drugs metabolized by cytochrome P450 enzymes, particularly those with a low therapeutic index.
Some cases of pancreatitis have been reported, which is paradoxical because SSAs are used to treat this disease.44
Dopamine agonistsThese were the first drugs to be specifically used for acromegaly. Because of their oral administration and lower costs, they are specially indicated for mild cases where GH and IGF-1 are slightly elevated.
Mechanism of actionDopamine agonists act through dopamine D2 receptors to decrease GH hypersecretion (Grade A). Bromocriptine was initially used, but only has some effectiveness in 10% of patients, and is therefore not recommended currently. Cabergoline, a more selective agonist, is the only dopamine agonist playing some role in the treatment of acromegaly.45
Dosage and administration schemesCabergoline (Dostinex®) is used at variable doses. The initial dose may be 1mg/week but, unlike for the treatment of prolactinoma, higher doses, up to 4mg/week in some cases, are required to achieve a normalization of biochemical changes.
The response should be monitored as for SSAs, by measuring GH and IGF-1 levels.
Clinical effectsDopamine agonists are less effective than SSAs and, as previously stated, are therefore more indicated for patients with mild IGF-1 elevations and for mixed tumors secreting GH and prolactin, although prolactin levels do not predict the response in acromegaly (Grade C).46
A recently published meta-analysis of 10 studies concluded that cabergoline monotherapy achieved a normalization of IGF-1 in one-third of the 170 patients analyzed. The available data regarding tumor size reduction are limited (Grade C).47
Adverse effects (Table 2)Cabergoline is better tolerated than bromocriptine (Grade C). However, it may cause gastrointestinal intolerance (nausea, vomiting, and epigastric discomfort), orthostatic hypotension, nasal congestion, dyspnea, and headache as the most common adverse effects, occurring in 10% of patients.48
Cardiac valve dysfunction may occur after the long-term use of these agents and when high doses are used (higher than 3mg/week of cabergoline). This adverse effect may occur in Parkinson's disease, where much higher doses are used, but has not been shown in acromegaly49,50 despite the fact that the dose may often be higher than that used in prolactinoma. Ultrasound monitoring is however recommended, particularly in patients on high cabergoline doses, and may be repeated every three years if normal.
Growth hormone receptor antagonist: pegvisomantMechanism of actionPegvisomant (PEG), the only GH antagonist, is a 199-amino acid pegylated product obtained by genetic recombination in Escherichia coli that behaves as a selective GH receptor antagonist. The substitution of glycine 120 in the third α helix chain of the GH molecule interferes with the second binding to the receptor. Eight additional amino acid substitutions increase its binding capacity in the first step, and pegylation confers on it a decreased antigenicity, increasing its half-life. Pegvisomant does not suppress dimerization, and the GHR complex is internalized, but is unable to produce an intracellular transduction signal. It does not decrease GH because it has no direct effect on the pituitary tumor.51,52
Dosage and administration schemesPEG is supplied as 10, 15, and 20mg vials for subcutaneous administration. PEG prescribing information recommends a starting dose of 80mg to achieve adequate concentrations, followed by doses ranging from 10 to 20mg/day, and this was the scheme used in clinical trials.53,54 Some patients may require up to 40mg/day. Because of its high costs, once or twice weekly administration schemes have been used, which may be helpful in some patients (Grade C).55
During treatment with PEG, IGF-1 levels should be monitored to assess efficacy (Grade B). It makes no sense to measure GH levels, which should increase but have no value for dosing (Grade B), because the results may also be altered by interference with the test method.
Clinical effectsPEG is the drug that best controls IGF-1 levels in acromegaly (Grade A), but its use is only currently approved in the event of SSA failure or when SSAs cannot be used because of their adverse effects.
Early clinical trials with pegvisomant administered at doses of 10, 15, or 20mg compared to placebo showed a 60% IGF-1 reduction at 12 weeks, with IGF-1 normalization in up to 89% of patients and in 97% of patients in a 18-month open label study, as well as dose-dependent decreases in symptoms (edema, joint pain) and signs (ring size, etc.) (Grade B).53,54
In the German Pegvisomant Observational Study, where 229 patients with acromegaly previously treated with surgery (90.4%), radiotherapy (43.2%), and medical therapy (94.3%) were given PEG, IGF-I normalization was achieved in 70.9% at 12 months and in 76.3% at 24 months.56
This lower efficacy in observational studies is probably due to inadequate adjustment of the doses used and to the fact that treatment with PEG has been indicated for patients refractory to other treatments.
In parallel to the IGF-1 decrease, an increase occurs in GH levels, which stabilize at 12–14ng/mL at six months of treatment. This is due to the interruption of negative feedback when IGF-1 production decreases.
Some factors may influence treatment response. Thus, it has been noted that a higher dose is required in women and patients with a higher body mass index, as well as prior GH and IGF-1 levels. By contrast, prior radiotherapy improves response.57
Unlike SSAs, various studies show that PEG improves glucose homeostasis, and may therefore be an option for diabetic or glucose intolerant patients (Grade C).58
Adverse effects (Table 2)A concern arising with PEG treatment is potential tumor growth due to the abovementioned loss of negative feedback, with a Nelson-like effect, as this drug has no antiproliferative activity.
Tumor growth has been reported in a few cases (3.2% in the German Pegvisomant Observational Study), but is probably related to the natural history of the tumor, because volume changes do not depend on treatment duration and are related to more aggressive tumors or to a rebound effect after SSA discontinuation (Grade C).59–61 Radiotherapy appears to decrease growth potential (Grade C), which has also been related to the duration of treatment with SSAs (Grade D).62
Monitoring with MRI every six months in the first year and annually thereafter is therefore recommended (Grade C), and PEG monotherapy should preferably be avoided in patients with big macroadenomas or tumors very close to the optic chiasm (Grade C).
Liver enzyme elevations three times above the normal range occur in 2.5% of patients treated with PEG, based on data from 1288 patients from the ACROSTUDY database63 (Grade B). These changes usually occur in the first three months of treatment, are usually self-limited, and are not related to dose (Grade B), but are related to prior SSA treatment64 (Grade D). Regular laboratory liver function tests and treatment discontinuation in patients with severe impairment or signs and symptoms of hepatitis are therefore recommended (Grade C).
Long-term treatment with PEG, because of its mechanism of action causes functional GH deficiency, and the aim of treatment should therefore be the maintenance of IGF-1 in the upper normal range, adjusted for age and sex.65
Other less common adverse effects of PEG include the development of hypertrophy at the injection site, which also usually occurs in the first weeks of treatment and resolves upon drug discontinuation.66 Allergic reactions, pain, and local erythema after injection, dizziness, headache, and flu-like syndrome more rarely occur (Grade C).67
Combination treatmentsIn some patients in whom optimal clinical or biochemical response is not achieved, a combination of the above drugs may be considered. Combination treatments are usually more effective and may allow for decreasing dosage and therefore toxicity, but another significant aspect to be considered is the economic cost.68,69
In patients not controlled on SSAs, the addition of cabergoline achieved a normalization of IGF-1 in 40% of patients in a study. The response was not related to hyperprolactinemia or immunohistochemistry positive for prolactin in the tumor (Grade C).70,71
Other recent studies assessed the combination of PEG with cabergoline.72,73 In the first such study, the addition of PEG at a fixed dose of 10mg to 0.5mg/day of cabergoline normalized IGF-1 levels in 68% of patients. When cabergoline was discontinued, only 26% continued to have normal IGF-1 levels.
The addition of PEG to SSAs is an attractive option because of their different and complementary mechanisms of action. In most studies, the combination appeared to be more effective for decreasing IGF-1 levels than did each drug acting alone,74 although the combination was not superior to PEG alone in a study on patients refractory to SSAs.75 It also allows for decreasing the frequency of PEG injection by one or two weekly doses.76
This combination has the advantage that SSAs may decrease tumor size and PEG may mitigate the harmful effect of SSAs on glucose homeostasis (Grade C). A potential disadvantage may be an increased liver toxicity with enzyme elevation, occurring in 11–15% of patients in the reported study, with a greater risk for diabetic patients.74
Larger studies are therefore needed to clarify combination treatment in the future.
A treatment algorithm mainly based on the specific characteristics of each patient is suggested below. Using the different treatment strategies, complete remission may currently be achieved in up to 95% of patients77 (Fig. 2).
RadiotherapyRadiotherapy is considered the third-line treatment for patients not controlled after surgery and those not responding to medical treatment (Grade C), although it may sometimes be considered the second-line treatment.8,9,78
Radiotherapy should be considered in patients not controlled after surgery in order to shorten the duration of medical treatment (Grade C).
Types of radiotherapyPatients amenable to radiotherapy should be given detailed information about its different modalities including its risks and benefits and side effects, as well as the need for long-term medical treatment, because disease control is usually delayed several years.
There are two modalities of radiotherapy: conventional conformational radiotherapy (CRT) and stereotactic radiotherapy (SRT), either as single (radiosurgery, RS) or as multiple doses (fractionated SRT, FSRT).78
CRT may decrease GH levels and normalize IGF-1 levels in 60% of patients, but maximal response may be achieved at 10–15 years. CRT achieves tumor volume control in 85–90% of patients, with tumor volume decrease in more than 50% of patients (Grade C). Tumor progression after radiotherapy is exceptional. Medical treatment should be continued while radiotherapy is effective (Grade B). It is usually administered at doses of 160–180cGy 4 or 5 days weekly for 5 or 6 weeks (total dose, 4500–5000cGy).
SRT techniques are currently of choice because they allow for better planning of the field to be irradiated, with a decreased risk of irradiation to adjacent structures.79,80
In patients with small tumor remnants, more than 5mm away from the optic tract and without excessively high GH and IGF-1 levels, treatment with CRT (gamma knife, linear accelerator, protons, etc.) is recommended (Grade C). CRT usually achieves earlier biochemical control. Remission rates have been reported to range from 17% to 50% in a two- to five-year follow-up period. Some series repeat CRT when remission is not achieved after the first dose. However, when such treatment should be repeated and the increase in complications which would warrant it have yet to be established.8,9,79–81
FSRT is advised in patients with large tumor remnants invading adjacent structures and high GH and IGF-1 levels, especially if they do not adequately respond to medical treatment.8,9,78,79
Medical treatment during radiotherapySome studies advise the discontinuation of medical treatment with SSAs or DAs at least one month before and during radiotherapy because of its radioprotective effect,82,83 but other studies do not support these findings.84 No conclusive data are available in this regard, and exactly when treatment should be discontinued before radiotherapy has not been established (Grade C).
MonitoringAfter radiotherapy, patients should continue medical treatment to achieve adequate control while radiotherapy is effective. Dose decrease and the periodic discontinuation of medical treatment is recommended to assess disease control (Grade B).
Pituitary function tests (gonadal, thyroid, and adrenal) should be performed annually to start replacement therapy if the patient has hypopituitarism (Grade B).9
ComplicationsLong-term hypopituitarism occurs in more than 50% of patients at five years (Grade A). Its incidence and severity is dose-dependent. In most series, the incidence of hypopituitarism is similar after RS and CRT. The presence of hypopituitarism is associated with increased mortality risk. In young patients who want to conceive, the risk of hypopituitarism should be considered before radiotherapy is recommended (Grade B).
Radiotherapy is also associated with an increased risk of mortality and cerebrovascular disease. The risk is dose-dependent. In a recent study by Sherlock et al. in 501 patients with acromegaly treated with radiotherapy (n=237), an increase was seen in all mortality causes as compared to non-irradiated patients (n=264). The risk of death from cerebrovascular disease was four times higher in irradiated patients. Vision loss occurred in some patients (0–3%) due to optic neuritis related to total cumulative dose.85
Radiotherapy was also associated with the occurrence of second tumors and radiation necrosis in 2% of patients treated with CRT. No data are available concerning the increased risks of mortality or cardiovascular disease in patients treated with RS. In a series of patients treated with RS, the incidence of radiation necrosis was 0.8%, although some patients had previously been treated with CRT. The risk of cognitive impairment after radiotherapy continues to be a controversial topic (Grade C).9
Comorbidities of acromegalyComorbid conditions associated with GH-secreting tumors may be due to local tumor effects, associated pituitary hormone deficiencies, or excess GH and IGF-1 levels. As regards comorbidities dependent on excess GH or IGF-1, the early detection and correction of associated risk factors facilitate a decrease in mortality secondary to these complications. Although the biochemical control of acromegaly may improve or stabilize these comorbidities, a significant proportion of patients require additional treatment.8,9,86,87
Cardiovascular complications and cardiovascular risk factorsArterial hypertensionArterial hypertension is highly prevalent (more than 40%) in patients with acromegaly, and blood pressure levels should therefore be routinely monitored. Arterial hypertension is aggravated by sleep apnea. Treatment for hypertension should be early and aggressive, independent of treatment for acromegaly, and with a high risk objective: to maintain blood pressure values at 130/80mmHg (Grade A, BEL 3). The specific treatment for arterial hypertension in these patients is not well defined.9,87
CardiomyopathyBiventricular hypertrophy is the typical finding of acromegalic heart disease. It is initially expressed as diastolic dysfunction on effort, which may progress to diastolic dysfunction at rest and, rarely, to dilated congestive cardiomyopathy. Arrhythmia may occur in 40% of patients.87
Assessment: cardiovascular risk assessment should include total cholesterol, LDL-C, HDL-C, and triglyceride levels (Grade C).9,87 Cardiological assessment should include an electrocardiogram. An echocardiogram is recommended if evidence of left ventricular hypertrophy is found in the electrocardiogram or symptoms including arrhythmia or dyspnea exist (Grade C).9,87
Patients with acromegaly, especially those with gigantism, require assessment of the peripheral artery system, including peripheral venous disease.
The potential effects of treatment for acromegaly: decreases in GH levels may improve left ventricular size and cardiac function (BEL 3). However, cardiac function normalizes in only 50% of patients older than 40 years.9,87
The potential valve effects of high-dose cabergoline in patients with acromegaly are controversial. However, an echocardiogram should be requested before cabergoline is started and at regular intervals thereafter.
The treatment of cardiac complications: standard treatment should be used to manage left ventricular hypertrophy, impaired diastolic or systolic function, arrhythmia, valve disorders, or ischemic disease (Grade C).
Diabetes mellitusDiabetes is a significant predictor of increased mortality in patients with acromegaly. Monitoring for and adequate treatment of diabetes should therefore be performed (Grade C). GH decrease usually improves glucose control, regardless of the treatment used.
Diabetes mellitus should be treated as in diabetic patients with no acromegaly in order to maintain hemoglobin A1c levels less than 6.5% (Grade C).9
In some patients in whom glucose control is impaired during treatment with SSAs, switching to PEG may be considered.9
Respiratory complicationsSleep apneaSleep apnea syndrome (SAS) is highly prevalent in acromegaly, affecting up to 70% of patients recently diagnosed. The main cause of SAS is obstruction due to pharyngeal thickening and macroglossia, although it may also have a central component. If obvious symptoms exist, home oximetry is recommended, followed by nocturnal polysomnography depending on the results (Grade C).9,87
The treatment of acromegaly decreases soft tissues and may induce SAS remission (EL 3), although SAS persistence is frequent. If this occurs, other treatment measures should be used, including assisted ventilation (CPAP) (for example, special measures for acromegaly such as specialized mouthpieces). Referral to a maxillofacial surgeon is also recommended (Grade C).9
Patients with upper respiratory tract obstruction due to jaw deformity, macroglossia, or enlarged epiglottis may experience complications during anesthesia. This should be considered before surgery (Grade C).9
Bone, joint, and dental complicationsBone and joint changes are not usually reversible after acromegaly has been cured (BEL 2). It is therefore advisable to delay any surgical procedure until GH and IFG-1 levels are normalized (Grade D).9
Joint diseaseDegeneration may affect any joint, and changes are usually irreversible (EL 3). Early diagnosis and treatment should be attempted, and aggressive management with physical therapy, systemic or intra-articular anti-inflammatory drugs, or prosthesis implantation when needed should be performed (Grade C).9
Carpian tunnel syndromeThe symptoms and/or signs of carpian tunnel syndrome should regularly be monitored. These symptoms may improve on GH reduction (EL 2), but if they persist, specific treatment should be performed, including surgery (Grade C9).
Hypercalciuria and hypercalcemiaThese may occur due to excess GH, are due to impaired vitamin D metabolism, and are reversible upon the cure of acromegaly (EL 3). If hypercalcemia persists, concomitant hyperthyroidism in the setting of multiple endocrine neoplasia should be ruled out.9
OsteoporosisPatients with acromegaly should be assessed for the risk factors of osteoporosis. Bone densitometry and the measurement of calcium levels are recommended at diagnosis, particularly if there is a history of hypogonadism or fracture. The value of DEXA as a diagnostic procedure for osteoporosis is not well documented in acromegalic patients, because increased bone thickness may influence bone mineral density measurement.
If osteoporosis exists and does not improve with the correction of hypogonadism or excess GH and IGF-1, antiresorptive treatment should be considered (Grade C).9
Polyps and colon cancerAlthough adequate evidence is not available for it to be possible to state that patients with acromegaly have a high risk of colon cancer, they do have a greater prevalence of colon polyps. A mortality rate from colon cancer greater than expected has also been seen (ratio 2.47). A screening colonoscopy is therefore recommended in adults at diagnosis (Grade C, BEL 3). If no polyps or cancer are found, monitoring as for the high-risk general population is recommended (a colonoscopy every five years). If the colonoscopy is abnormal, specific follow-up clinical guidelines should be followed.9,87
Psychosocial complicationsAcromegaly is associated with an impaired quality of life (EL 3), and specific tests to measure this variable, such as AcroQoL, are an important tool for measuring the results and should be used in clinical practice.
Biochemical control improves several aspects of psychosocial functioning (EL 3), although biochemical normalization does not always normalize these aspects.9
MortalityAcromegaly is associated with a 2- to 2.5-fold increase in mortality. GH of IGF-1 normalization may improve the mortality risk (BEL 2).88 GH levels (measured using sensitive methods) less than 1 are associated with a mortality rate similar to what would normally be expected (EL 2).
Long-term monitoring of acromegalyThis should be performed based on the degree of disease control (Table 3):
- 1.
Acromegaly cured after surgery: once postoperative control is confirmed (3–6 months after surgery) by GH and IGF-1 normalization and GH suppression after OGTT (below 1 or 0.4μg/L), patients should be monitored regularly. Serial measurements of GH and IGF-1 levels should be performed (every 6 months initially and annually thereafter). If recurrence criteria are found, OGTT should be performed for confirmation.
- 2.
Acromegaly controlled with medical treatment: patients treated with SSAs should be monitored with measurements of GH and IGF-1, initially every three or four months for dose adjustment, and biannually or annually thereafter. OGTT is not required during monitoring. After adequate control, the SSA dose interval may be lengthened or the dose may be decreased. In patients treated with PEG, monitoring should only consist of IGF-1 measurement.
- 3.
Acromegaly treated with radiotherapy: monitoring should be the same as for patients on medical treatment. After adequate control, the drug dose should be decreased or discontinued to confirm the definitive cure. Pituitary function should also be regularly monitored for hypopituitarism.
MRI should be performed three or four months after surgery or after the start of medical treatment. Subsequent monitoring will depend on disease control. If the patient is cured after surgery, no additional MRIs will be needed, although this is not clearly established. An alternative could be to perform MRI every two or three years, or sooner if clinical or biochemical recurrence occurs. In patients controlled on SSAs, MRI should be performed at one year to verify tumor size decrease. The subsequent frequency of MRI is not established and will depend on clinical judgment. If the patient is not controlled, MRI should be performed every six months initially and annually thereafter. In patients treated with PEG, MRI should be performed every six months initially to check for tumor growth and annually thereafter. In patients treated with radiotherapy, annual monitoring should be performed until tumor disappearance or control.
An important aspect of long-term monitoring is the control of comorbidities and, in irradiated patients, the monitoring of pituitary function in order that replacement therapy may be started if hypopituitarism occurs.
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Cordido F, García Arnés JA, Marazuela Aspiroz M, Torres Vela E, en representación del grupo de Neuroendocrinología de la Sociedad Española de Endocrinología y Nutrición. Guía práctica de diagnóstico y tratamiento de la acromegalia. Endocrinol Nutr. 2013;60:457.
